
BiteSized Immunology: Systems & Processes

Antigen Processing and Presentation
In order to be capable of engaging the key elements of adaptive immunity (specificity, memory, diversity, self/nonself discrimination), antigens have to be processed and presented to immune cells. Antigen presentation is mediated by MHC class I molecules , and the class II molecules found on the surface of antigen-presenting cells (APCs) and certain other cells.
MHC class I and class II molecules are similar in function: they deliver short peptides to the cell surface allowing these peptides to be recognised by CD8+ (cytotoxic) and CD4+ (helper) T cells, respectively. The difference is that the peptides originate from different sources – endogenous, or intracellular , for MHC class I; and exogenous, or extracellular for MHC class II. There is also so called cross-presentation in which exogenous antigens can be presented by MHC class I molecules. Endogenous antigens can also be presented by MHC class II when they are degraded through autophagy.

MHC class I presentation
MHC class I molecules are expressed by all nucleated cells. MHC class I molecules are assembled in the endoplasmic reticulum (ER) and consist of two types of chain – a polymorphic heavy chain and a chain called β2-microglobulin. The heavy chain is stabilised by the chaperone calnexin , prior to association with the β2-microglobulin. Without peptides, these molecules are stabilised by chaperone proteins : calreticulin, Erp57, protein disulfide isomerase (PDI) and tapasin. The complex of TAP, tapasin, MHC class I, ERp57 and calreticulin is called the peptide-loading complex (PLC). Tapasin interacts with the transport protein TAP (transporter associated with antigen presentation) which translocates peptides from the cytoplasm into the ER. Prior to entering the ER, peptides are derived from the degradation of proteins, which can be of viral- or self origin. Degradation of proteins is mediated by cytosolic- and nuclear proteasomes, and the resulting peptides are translocated into the ER by means of TAP. TAP translocates peptides of 8 –16 amino acids and they may require additional trimming in the ER before binding to MHC class I molecules. This is possibly due to the presence of ER aminopeptidase (ERAAP) associated with antigen processing.
It should be noted that 30–70% of proteins are immediately degraded after synthesis (they are called DRiPs – defective ribosomal products, and they are the result of defective transcription or translation). This process allows viral peptides to be presented very quickly – for example, influenza virus can be recognised by T cells approximately 1.5 hours post-infection. When peptides bind to MHC class I molecules, the chaperones are released and peptide–MHC class I complexes leave the ER for presentation at the cell surface. In some cases, peptides fail to associate with MHC class I and they have to be returned to the cytosol for degradation. Some MHC class I molecules never bind peptides and they are also degraded by the ER-associated protein degradation (ERAD) system.
There are different proteasomes that generate peptides for MHC class-I presentation: 26S proteasome , which is expressed by most cells; the immunoproteasome, which is expressed by many immune cells; and the thymic-specific proteasome expressed by thymic epithelial cells.
Antigen presentation
On the surface of a single cell, MHC class I molecules provide a readout of the expression level of up to 10,000 proteins. This array is interpreted by cytotoxic T lymphocytes and Natural Killer cells, allowing them to monitor the events inside the cell and detect infection and tumorigenesis.
MHC class I complexes at the cell surface may dissociate as time passes and the heavy chain can be internalised. When MHC class I molecules are internalised into the endosome, they enter the MHC class-II presentation pathway. Some of the MHC class I molecules can be recycled and present endosomal peptides as a part of a process which is called cross-presentation .
The usual process of antigen presentation through the MHC I molecule is based on an interaction between the T-cell receptor and a peptide bound to the MHC class I molecule. There is also an interaction between the CD8+ molecule on the surface of the T cell and non-peptide binding regions on the MHC class I molecule. Thus, peptide presented in complex with MHC class I can only be recognised by CD8+ T cells. This interaction is a part of so-called ‘three-signal activation model’, and actually represents the first signal. The next signal is the interaction between CD80/86 on the APC and CD28 on the surface of the T cell, followed by a third signal – the production of cytokines by the APC which fully activates the T cell to provide a specific response.
MHC class I polymorphism
Human MHC class I molecules are encoded by a series of genes – HLA-A, HLA-B and HLA-C (HLA stands for ‘Human Leukocyte Antigen’, which is the human equivalent of MHC molecules found in most vertebrates). These genes are highly polymorphic, which means that each individual has his/her own HLA allele set. The consequences of these polymorphisms are differential susceptibilities to infection and autoimmune diseases that may result from the high diversity of peptides that can bind to MHC class I in different individuals. Also, MHC class I polymorphisms make it virtually impossible to have a perfect tissue match between donor and recipient, and thus are responsible for graft rejection.

MHC class II presentation
MHC class II molecules are expressed by APCs, such as dendritic cells (DC), macrophages and B cells (and, under IFNγ stimuli, by mesenchymal stromal cells, fibroblasts and endothelial cells, as well as by epithelial cells and enteric glial cells). MHC class II molecules bind to peptides that are derived from proteins degraded in the endocytic pathway. MHC class II complexes consists of α- and β-chains that are assembled in the ER and are stabilised by invariant chain (Ii). The complex of MHC class II and Ii is transported through the Golgi into a compartment which is termed the MHC class II compartment (MIIC). Due to acidic pH, proteases cathepsin S and cathepsin L are activated and digest Ii, leaving a residual class II-associated Ii peptide (CLIP) in the peptide-binding groove of the MHC class II. Later, the CLIP is exchanged for an antigenic peptide derived from a protein degraded in the endosomal pathway. This process requires the chaperone HLA-DM, and, in the case of B cells, the HLA-DO molecule. MHC class II molecules loaded with foreign peptide are then transported to the cell membrane to present their cargo to CD4+ T cells. Thereafter, the process of antigen presentation by means of MHC class II molecules basically follows the same pattern as for MHC class I presentation.
As opposed to MHC class I, MHC class II molecules do not dissociate at the plasma membrane. The mechanisms that control MHC class II degradation have not been established yet, but MHC class II molecules can be ubiquitinised and then internalised in an endocytic pathway.
MHC class II polymorphism
Like the MHC class I heavy chain, human MHC class II molecules are encoded by three polymorphic genes: HLA-DR, HLA-DQ and HLA-DP. Different MHC class II alleles can be used as genetic markers for several autoimmune diseases, possibly owing to the peptides that they present.

- DNA Replication
- Active Transport
- Cellular Receptors
- Endocytosis and Exocytosis
- Enzyme Inhibition
- Enzyme Kinetics
- Protein Structure
- Transcription of DNA
- Translation of DNA
- Anaerobic Respiration
- Electron Transport Chain
- Gluconeogenesis
- Calcium Regulation
- External Balance of Potassium
- Internal Balance of Potassium
- Sodium Regulation
- Cell Membrane
- Endoplasmic Reticulum
- Golgi Apparatus
- Mitochondria
- Blood Vessels
- Cellular Adaptations
- Epithelial Cells
- Muscle Histology
- Structure of Glands
- Control of Stroke Volume
- Control of Heart Rate
- Cardiac Cycle
- Cardiac Pacemaker Cells
- Conduction System
- Contraction of Cardiac Muscle
- Ventricular Action Potentials
- Blood Flow in Vessels
- Control of Blood Pressure
- Capillary Exchange
- Flow In Peripheral Circulation
- Venous Return
- Cardiac Muscle
- Hepatic Circulation
- Skeletal Muscle
- Airway Resistance
- Lung Volumes
- Mechanics of Breathing
- Gas Exchange
- Oxygen Transport in The Blood
- Transport of Carbon Dioxide in the Blood
- Ventilation-Perfusion Matching
- Chemoreceptors
- Cough Reflex
- Neural Control of Ventilation
- Respiratory Regulation of Acid-Base Balance
- Responses of The Respiratory System to Stress
- Regulation of Saliva
- Secretion of Saliva
- Gastric Acid Production
- Gastric Mucus Production
- Digestion and Absorption
- Histology and Cellular Function of the Small Intestine
- Absorption in the Large Intestine
- Large Intestinal Motility
- Bilirubin Metabolism
- Carbohydrate Metabolism in the Liver
- Lipid Metabolism in the Liver
- Protein and Ammonia Metabolism in the Liver
- Storage Functions of the Liver
- Bile Production
- Function of The Spleen
- Exocrine Pancreas
- Somatostatin
- Proximal Convoluted Tubule
- Loop of Henle
- Distal Convoluted Tubule and Collecting Duct
- Storage Phase of Micturition
- Voiding Phase of Micturition
- Antidiuretic Hormone
- Renin-Angiotensin-Aldosterone System
- Urinary Regulation of Acid-Base Balance
- Water Filtration and Reabsorption
- Development of the Reproductive System
- Gametogenesis
- Gonadotropins and the Hypothalamic Pituitary Axis
- Menstrual Cycle
- Placental Development
- Fetal Circulation
- Maternal Adaptations in Pregnancy
- Cells of the Nervous System
- Central Nervous System
- Cerebrospinal Fluid
- Neurotransmitters
- Peripheral Nervous System
- Action Potential
- Excitatory and Inhibitory Synaptic Signalling
- Resting Membrane Potential
- Synaptic Plasticity
- Synaptic Transmission
- Ascending Tracts
- Auditory Pathway
- Consciousness and Sleep
- Modalities of Sensation
- Pain Pathways
- Sensory Acuity
- Visual Pathway
- Descending Tracts
- Lower Motor Neurones
- Muscle Stretch Reflex
- Upper Motor Neurones
- Aqueous Humour
- Ocular Accommodation
- Thyroid Gland
- Parathyroid Glands
- Adrenal Medulla
- Zona Glomerulosa
- Zona Fasciculata
- Zona Reticularis
- Endocrine Pancreas
- The Hypothalamus
- Anterior Pituitary
- Posterior Pituitary
- White Blood Cells – Summary
- Barriers to Infection
- Infection Recognition Molecules
- Phagocytosis
- The Complement System
Antigen Processing and Presentation
- Primary and Secondary Immune Responses
- T Cell Memory
- Acute Inflammation
- Autoimmunity
- Chronic Inflammation
- Hypersensitivity Reactions
- Immunodeficiency
- Types of Immunity
- Antibiotics
- Viral Infection
- Blood Groups
- Coagulation
- Erythropoiesis
- Iron Metabolism
- Mononuclear Phagocyte System
Original Author(s): Antonia Round Last updated: 17th July 2023 Revisions: 9
- 1 Antigen Presentation
- 2.1 MHC Class I Molecules
- 2.2 MCH Class II Molecules
- 3.1 T Cell Receptors
- 3.2 Co-Receptors
- 4 Clinical Relevance – Autoimmune disease
T cells can only recognise antigens when they are displayed on cell surfaces. This is carried out by Antigen-presenting cells (APCs) , the most important of which are dendritic cells, B cells, and macrophages. APCs can digest proteins they encounter and display peptide fragments from them on their surfaces for other immune cells to recognise.
This process of antigen presentation allows T cells to “see” what proteins are present in the body and to form an adaptive immune response against them. In this article, we shall discuss antigen processing, presentation, and recognition by T cells.
Antigen Presentation
Antigens are delivered to the surface of APCs by Major Histocompatibility Complex (MHC) molecules. Different MHC molecules can bind different peptides. The MHC is highly polygenic and polymorphic which equips us to recognise a vast array of different antigens we might encounter. There are different classes of MHC, which have different functions:
- MHC class I molecules are found on all nucleated cells (not just professional APCs) and typically present intracellular antigens such as viruses.
- MHC class II molecules are only found on APCs and typically present extracellular antigens such as bacteria.
This is logical because should a virus be inside a cell of any type, the immune system needs to be able to respond to it. This also explains why pathogens inside human red blood cells (which are non-nucleated) can be difficult for the immune system to find, such as in malaria.
Whilst this is the general rule, in cross-presentation extracellular antigens can be presented by MHC class I, and in autophagy intracellular antigens can be presented by MHC class II.
Antigen Processing
Before an antigen can be presented, it must first be processed . Processing transforms proteins into antigenic peptides.
MHC Class I Molecules
Intracellular peptides for MHC class I presentation are made by proteases and the proteasome in the cytosol, then transported into the endoplasmic reticulum via TAP (Transporter associated with Antigen Processing) to be further processed.
They are then assembled together with MHC I molecules and travel to the cell surface ready for presentation.
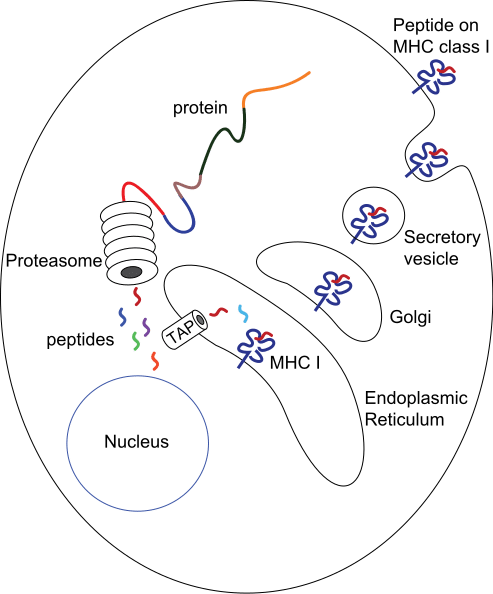
Fig 1 – Diagram demonstrating the production of peptides for MHC class I presentation
MCH Class II Molecules
The route of processing for exogenous antigens for MHC class II presentation begins with endocytosis of the antigen. Once inside the cell, they are encased within endosomes that acidify and activate proteases, to degrade the antigen.
MHC class II molecules are transported into endocytic vesicles where they bind peptide antigen and then travel to the cell surface.
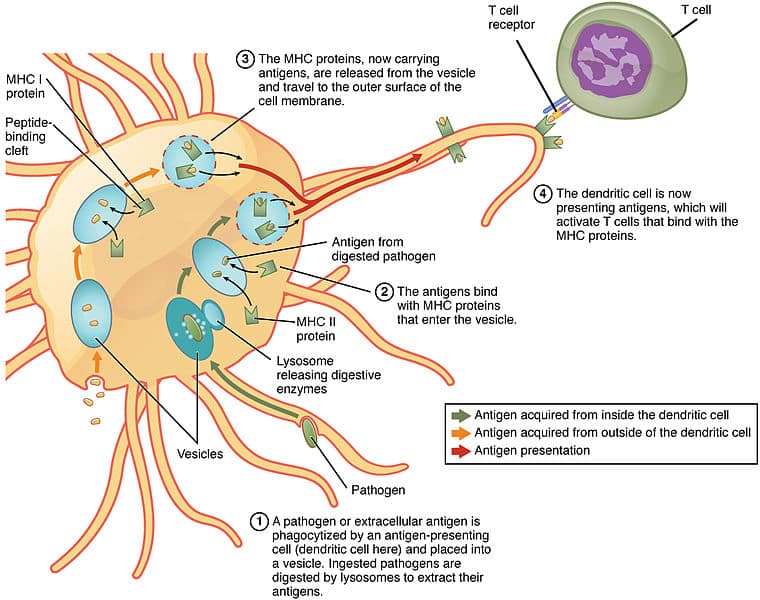
Fig 2 – Diagram showing processing of antigens for MHC Class II presentation by a dendritic cell
The antigen presented on MHCs is recognised by T cells using a T cell receptor (TCR) . These are antigen-specific .
T Cell Receptors
Each T cell has thousands of TCRs , each with a unique specificity that collectively allows our immune system to recognise a wide array of antigens.
This diversity in TCRs is achieved through a process called V(D)J recombination during development in the thymus. TCR chains have a variable region where gene segments are randomly rearranged, using the proteins RAG1 and RAG2 to initiate cleavage and non-homologous end joining to rejoin the chains.
The diversity of the TCRs can be further increased by inserting or deleting nucleotides at the junctions of gene segments; together forming the potential to create up to 10 15 unique TCRs.
TCRs are specific not only for a particular antigen but also for a specific MHC molecule. T cells will only recognise an antigen if a specific antigen with a specific MHC molecule is present: this phenomenon is called MHC restriction .
Co-Receptors
As well as the TCR, another T cell molecule is required for antigen recognition and is known as a co-receptor. These are either a CD4 or CD8 molecule:
- CD4 is present on T helper cells and only binds to antigen-MHC II complexes.
- CD8 is present on cytotoxic T cells and only binds to antigen-MHC I complexes.
This, therefore, leads to very different effects. Antigens presented with MHC II will activate T helper cells and antigens presented with MHC I activate cytotoxic T cells. Cytotoxic T cells will kill the cells that they recognise, whereas T helper cells have a broader range of effects on the presenting cell such as activation to produce antibodies (in the case of B cells) or activation of macrophages to kill their intracellular pathogens.
Clinical Relevance – Autoimmune disease
It is important to note that APCs may deliver foreign antigens or self-antigens. In the case of autoimmune diseases, self-antigens are presented to T cells, which then initiates an immune response against our own tissues.
For example, in Graves’ disease , TSHR (thyroid stimulating hormone receptor) acts as a self-antigen and is presented to T cells. This then activates B cells to produce autoantibodies against TSHRs in the thyroid. This results in the activation of TSHRs leading to hyperthyroidism and a possible goitre.
[start-clinical]
Clinical Relevance - Autoimmune disease
[end-clinical]
Found an error? Is our article missing some key information? Make the changes yourself here!
Once you've finished editing, click 'Submit for Review', and your changes will be reviewed by our team before publishing on the site.
We use cookies to improve your experience on our site and to show you relevant advertising. To find out more, read our privacy policy .
Privacy Overview

- school Campus Bookshelves
- menu_book Bookshelves
- perm_media Learning Objects
- login Login
- how_to_reg Request Instructor Account
- hub Instructor Commons
Margin Size
- Download Page (PDF)
- Download Full Book (PDF)
- Periodic Table
- Physics Constants
- Scientific Calculator
- Reference & Cite
- Tools expand_more
- Readability
selected template will load here
This action is not available.
20.3E: Antigen-Presenting Cells
- Last updated
- Save as PDF
- Page ID 7949
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
Antigen presentation is a process by which immune cells capture antigens and then enable their recognition by T cells.
Learning Objectives
- Describe the role of antigen-presenting cells
- The host’s cells express “self” antigens that identify them as such. These antigens are different from those in bacteria (“non-self” antigens) and in virus-infected host cells (“missing-self”).
- Antigen presentation consists of pathogen recognition, phagocytosis of the pathogen or its molecular components, processing of the antigen, and then presentation of the antigen to naive T cells.
- The T cell receptor is restricted to recognizing antigenic peptides only when bound to appropriate molecules of the major histocompatibility complex (MHC), also known in humans as human leukocyte antigen (HLA).
- Helper T cells recieve antigens from MHC II on an APC, while cytotoxic T cells recieve antigens from MHC I. Helper T cells present their antigen to B cells as well.Dendritic cells, B cells, and macrophages play a major role in the innate response, and are the primary antigen-presenting cells (APC).
- APCs use toll-like receptors to identify PAMPS and DAMPs, which are signs of an infection and may be processed into antigen peptides if phagocytized. Most APCs cannot tell the difference between different types of antigens like B and T cells can.
- damage-associated molecular pattern : Protein or nucleic acid based signs of pathogen induced damage. Protein DAMPs may be phagocytized and processed for antigen presentation.
- cytotoxic : A population of T cells specialized for inducing the deaths of other cells.
Antigen presentation is a process in the body’s immune system by which macrophages, dendritic cells and other cell types capture antigens, then present them to naive T-cells. The basis of adaptive immunity lies in the capacity of immune cells to distinguish between the body’s own cells and infectious pathogens. The host’s cells express “self” antigens that identify them as belonging to the self. These antigens are different from those in bacteria (“non-self” antigens) or in virally-infected host cells (“missing-self”). Antigen presentation broadly consists of pathogen recognition, phagocytosis of the pathogen or its molecular components, processing of the antigen, and then presentation of the antigen to naive (mature but not yet activated) T cells. The ability of the adaptive immune system to fight off pathogens and end an infection depends on antigen presentation.
Antigen Presenting Cells
Antigen Presenting Cells (APCs) are cells that capture antigens from within the body, and present them to naive T-cells. Many immune system cells can present antigens, but the most common types are macrophages and dendritic cells, which are two types of terminally differentiated leukocytes that arise from monocytes. Both of these APCs perform many immune functions that are important for both innate and adaptive immunity, such as removing leftover pathogens and dead neutrophils after an inflammatory response. Dendritic cells (DCs) are generally found in tissues that have contact with the external environment (such as the skin or respiratory epithelium) while macrophages are found in almost all tissues. Some types of B cells may also present antigens as well, though it is not their primary function.
APCs phagocytize exogenous pathogens such as bacteria, parasites, and toxins in the tissues and then migrate, via chemokine signals, to lymph nodes that contain naive T cells. During migration, APCs undergo a process of maturation in which they digest phagocytized pathogens and begin to express the antigen in the form of a peptide on their MHC complexes, which enables them to present the antigen to naive T cells. The antigen digestion phase is also called “antigen processing,” because it prepares the antigens for presentation. This MHC:antigen complex is then recognized by T cells passing through the lymph node. Exogenous antigens are usually displayed on MHC Class II molecules, which interact with CD4+ helper T cells.
This maturation process is dependent on signaling from other pathogen-associated molecular pattern (PAMP) molecules (such as a toxin or component of a cell membrane from a pathogen) through pattern recognition receptors (PRRs), which are received by Toll-like receptors on the DC’s body. They may also recognize damage-associated molecular pattern (DAMP) molecules, which include degraded proteins or nucleic acids released from cells that undergo necrosis. PAMPs and DAMPS are not technically considered antigens themselves, but instead are signs of pathogen presence that alert APCs through Toll-like receptor binding. However if a DC phagocytzes a PAMP or DAMP, it could be used as an antigen during antigen presentation. APCs are unable to distinguish between different types of antigens themselves, but B and T cells can due to their specificity.
Antigen Presentation
T cells must be presented with antigens in order to perform immune system functions. The T cell receptor is restricted to recognizing antigenic peptides only when bound to appropriate molecules of the MHC complexes on APCs, also known in humans as Human leukocyte antigen (HLA).
Several different types of T cell can be activated by APCs, and each type of T cell is specially equipped to deal with different pathogens, whether the pathogen is bacterial, viral or a toxin. The type of T cell activated, and therefore the type of response generated, depends on which MHC complex the processed antigen-peptide binds to.
MHC Class I molecules present antigen to CD8+ cytotoxic T cells, while MHC class II molecules present antigen to CD4+ helper T cells. With the exception of some cell types (such as erythrocytes), Class I MHC is expressed by almost all host cells. Cytotoxic T cells (also known as TC, killer T cell, or cytotoxic T-lymphocyte (CTL)) are a population of T cells that are specialized for inducing the death of other cells. Recognition of antigenic peptides through Class I by CTLs leads to the killing of the target cell, which is infected by virus, intracytoplasmic bacterium, or are otherwise damaged or dysfunctional. Additionally, some helper T cells will present their antigen to B cells, which will activate their proliferation response.
Antigen presentation : In the upper pathway; foreign protein or antigen (1) is taken up by an antigen-presenting cell (2). The antigen is processed and displayed on a MHC II molecule (3), which interacts with a T helper cell (4). In the lower pathway; whole foreign proteins are bound by membrane antibodies (5) and presented to B lymphocytes (6), which process (7) and present antigen on MHC II (8) to a previously activated T helper cell (10), spurring the production of antigen-specific antibodies (9).
LICENSES AND ATTRIBUTIONS
CC LICENSED CONTENT, SHARED PREVIOUSLY
- Curation and Revision. Authored by : Boundless.com. Provided by : Boundless.com. License : CC BY-SA: Attribution-ShareAlike
CC LICENSED CONTENT, SPECIFIC ATTRIBUTION
- antigen. Provided by : Wiktionary. Located at : en.wiktionary.org/wiki/antigen . License : CC BY-SA: Attribution-ShareAlike
- Adaptive immune system. Provided by : Wikipedia. Located at : en.Wikipedia.org/wiki/Adaptive_immune_system . License : CC BY-SA: Attribution-ShareAlike
- antibody. Provided by : Wiktionary. Located at : en.wiktionary.org/wiki/antibody . License : CC BY-SA: Attribution-ShareAlike
- macrophage. Provided by : Wiktionary. Located at : en.wiktionary.org/wiki/macrophage . License : CC BY-SA: Attribution-ShareAlike
- Antibody. Provided by : Wikipedia. Located at : en.Wikipedia.org/wiki/File:Antibody.jpg . License : Public Domain: No Known Copyright
- Antigen presentation. Provided by : Wikipedia. Located at : en.Wikipedia.org/wiki/File:Antigen_presentation.svg . License : Public Domain: No Known Copyright
- Immune system. Provided by : Wikipedia. Located at : en.Wikipedia.org/wiki/Immune_system%23Innate_immune_system . License : CC BY-SA: Attribution-ShareAlike
- Lymphocyte_activation_simple.png. Provided by : Wikipedia. Located at : en.Wikipedia.org/wiki/Immune_system#/media/File:Lymphocyte_activation_simple.png . License : CC BY-SA: Attribution-ShareAlike
- T cell. Provided by : Wikipedia. Located at : en.Wikipedia.org/wiki/T_cell . License : CC BY-SA: Attribution-ShareAlike
- B cell. Provided by : Wikipedia. Located at : en.Wikipedia.org/wiki/B_cell . License : CC BY-SA: Attribution-ShareAlike
- Immune cells. Provided by : Wikipedia. Located at : en.Wikipedia.org/wiki/Immune_cells . License : CC BY-SA: Attribution-ShareAlike
- thymus. Provided by : Wiktionary. Located at : en.wiktionary.org/wiki/thymus . License : CC BY-SA: Attribution-ShareAlike
- Red White Blood cells. Provided by : Wikipedia. Located at : en.Wikipedia.org/wiki/File:Red_White_Blood_cells.jpg . License : Public Domain: No Known Copyright
- Lymphocyte. Provided by : Wikipedia. Located at : en.Wikipedia.org/wiki/Lymphocyte . License : CC BY-SA: Attribution-ShareAlike
- B cells. Provided by : Wikipedia. Located at : en.Wikipedia.org/wiki/B%20cells . License : CC BY-SA: Attribution-ShareAlike
- natural killer (NK) cells. Provided by : Wikipedia. Located at : en.Wikipedia.org/wiki/natural...20(NK)%20cells . License : CC BY-SA: Attribution-ShareAlike
- T cells. Provided by : Wiktionary. Located at : en.wiktionary.org/wiki/T_cells . License : CC BY-SA: Attribution-ShareAlike
- Lymphocyte. Provided by : Wikipedia. Located at : en.Wikipedia.org/wiki/Lymphocyte . License : Public Domain: No Known Copyright
- Antigen presentation. Provided by : Wikipedia. Located at : en.Wikipedia.org/wiki/Antigen_presentation . License : CC BY-SA: Attribution-ShareAlike
- cytotoxic. Provided by : Wiktionary. Located at : en.wiktionary.org/wiki/cytotoxic . License : CC BY-SA: Attribution-ShareAlike
- Antigen presentation. Provided by : Wikipedia. Located at : en.Wikipedia.org/wiki/Antigen_presentation . License : Public Domain: No Known Copyright
Module 20: The Immune System
Antigen-presenting cells, learning outcomes.
- Describe the structure and function of antigen-presenting cells
Unlike NK cells of the innate immune system, B cells (B lymphocytes) are a type of white blood cell that gives rise to antibodies, whereas T cells (T lymphocytes) are a type of white blood cell that plays an important role in the immune response. T cells are a key component in the cell-mediated response—the specific immune response that utilizes T cells to neutralize cells that have been infected with viruses and certain bacteria. There are three types of T cells: cytotoxic, helper, and suppressor T cells. Cytotoxic T cells destroy virus-infected cells in the cell-mediated immune response, and helper T cells play a part in activating both the antibody and the cell-mediated immune responses. Suppressor T cells deactivate T cells and B cells when needed, and thus prevent the immune response from becoming too intense.
An antigen is a foreign or “non-self” macromolecule that reacts with cells of the immune system. Not all antigens will provoke a response. For instance, individuals produce innumerable “self” antigens and are constantly exposed to harmless foreign antigens, such as food proteins, pollen, or dust components. The suppression of immune responses to harmless macromolecules is highly regulated and typically prevents processes that could be damaging to the host, known as tolerance.
The innate immune system contains cells that detect potentially harmful antigens, and then inform the adaptive immune response about the presence of these antigens. An antigen-presenting cell (APC) is an immune cell that detects, engulfs, and informs the adaptive immune response about an infection. When a pathogen is detected, these APCs will phagocytose the pathogen and digest it to form many different fragments of the antigen. Antigen fragments will then be transported to the surface of the APC, where they will serve as an indicator to other immune cells. Dendritic cells are immune cells that process antigen material; they are present in the skin (Langerhans cells) and the lining of the nose, lungs, stomach, and intestines. Sometimes a dendritic cell presents on the surface of other cells to induce an immune response, thus functioning as an antigen-presenting cell. Macrophages also function as APCs. Before activation and differentiation, B cells can also function as APCs.
After phagocytosis by APCs, the phagocytic vesicle fuses with an intracellular lysosome forming phagolysosome. Within the phagolysosome, the components are broken down into fragments; the fragments are then loaded onto MHC class I or MHC class II molecules and are transported to the cell surface for antigen presentation, as illustrated in Figure 1. Note that T lymphocytes cannot properly respond to the antigen unless it is processed and embedded in an MHC II molecule. APCs express MHC on their surfaces, and when combined with a foreign antigen, these complexes signal a “non-self” invader. Once the fragment of antigen is embedded in the MHC II molecule, the immune cell can respond. Helper T- cells are one of the main lymphocytes that respond to antigen-presenting cells. Recall that all other nucleated cells of the body expressed MHC I molecules, which signal “healthy” or “normal.”

Figure 1. An APC, such as a macrophage, engulfs and digests a foreign bacterium. An antigen from the bacterium is presented on the cell surface in conjunction with an MHC II molecule Lymphocytes of the adaptive immune response interact with antigen-embedded MHC II molecules to mature into functional immune cells.
- Biology 2e. Provided by : OpenStax. Located at : http://cnx.org/contents/[email protected] . License : CC BY: Attribution . License Terms : Access for free at https://openstax.org/books/biology-2e/pages/1-introduction


- school Campus Bookshelves
- menu_book Bookshelves
- perm_media Learning Objects
- login Login
- how_to_reg Request Instructor Account
- hub Instructor Commons
Margin Size
- Download Page (PDF)
- Download Full Book (PDF)
- Periodic Table
- Physics Constants
- Scientific Calculator
- Reference & Cite
- Tools expand_more
- Readability
selected template will load here
This action is not available.
15.4M: Antigen Presentation
- Last updated
- Save as PDF
- Page ID 5449

- John W. Kimball
- Tufts University & Harvard
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
Antigens are macromolecules that elicit an immune response in the body. Antigens can be proteins, polysaccharides, conjugates of lipids with proteins (lipoproteins) and polysaccharides (glycolipids). Most of this page will describe how protein antigens are presented to the immune system. The presentation of lipid and polysaccharide antigens will be mentioned at the end. It will be helpful to distinguish between two limiting cases.
Antigens that enter the body from the environment; these would include inhaled macromolecules (e.g., proteins on cat hairs that can trigger an attack of asthma in susceptible people), ingested macromolecules (e.g., shellfish proteins that trigger an allergic response in susceptible people), and molecules that are introduced beneath the skin (e.g., on a splinter or in an injected vaccine). Alternatively, antigens can be generated within the cells of the body; these would include proteins encoded by the genes of viruses that have infected a cell and aberrant proteins that are encoded by mutant genes; such as mutated genes in cancer cells. In all cases, however, the initial immune response to any antigen absolutely requires that the antigen be recognized by a T lymphocyte ("T cell"). The truth of this rule is clearly demonstrated in AIDS : the infections (viral or fungal or bacterial) that so often claim the life of AIDS patients do so when the patient has lost virtually all of his or her CD4 + T cells. The two categories of antigens are processed and presented to T cells by quite different mechanisms.
Exogenous Antigens
Exogenous antigens (inhaled, ingested, or injected) are taken up by antigen-presenting cells (APCs). These include phagocytic cells like dendritic cells and macrophages and B lymphocytes ("B cells") which are responsible for producing antibodies against the antigen. Antigen-presenting cells
- engulf the antigen by endocytosis
- endosome fuses with a lysosome where the antigen is degraded into fragments (e.g. short peptides)
- these antigenic peptides are then displayed at the surface of the cell nestled within a class II histocompatibility molecule .
- they may be recognized by CD4 + T cells
The Class I Pathway
Class I histocompatibility molecules are transmembrane proteins expressed at the cell surface. Like all transmembrane proteins, they are synthesized by ribosomes on the rough endoplasmic reticulum (RER) and assembled within its lumen. There are three subunits in each class I histocompatibility molecule:
- the transmembrane polypeptide (called the "heavy chain")
- the antigenic peptide
- beta-2 microglobulin
All of these must be present within the lumen of the endoplasmic reticulum if they are to assemble correctly and move through the Golgi apparatus to the cell surface. The Problem: proteins encoded by the genes of an infecting virus are synthesized in the cytosol . How to get them into the endoplasmic reticulum?
The Solution: TAP (= t ransporter a ssociated with antigen p rocessing).
- Viral proteins in the cytosol are degraded by proteasomes into viral peptides.
- The peptides are picked up by TAP proteins embedded in the membrane of the endoplasmic reticulum.
- Using the energy of ATP, the peptides are pumped into the lumen of the endoplasmic reticulum where they assemble with the transmembrane polypeptide and beta-2 microglobulin.
- This trimolecular complex then moves through the Golgi apparatus and is inserted in the plasma membrane.
- The complex can be bound by a T cell with a receptor ( TCR ) able to bind the peptide and flanking portions of the histocompatibility molecule (the hot dog in the bun) and CD8 molecules that bind the CD8 receptor (shown above as a gray hemisphere) on the histocompatibility molecule.
The Class II Pathway
Class II histocompatibility molecules consist of two transmembrane polypeptides and a third molecule nestled in the groove they form. All three components of this complex must be present in the endoplasmic reticulum for proper assembly. But antigenic peptides are not transported to the endoplasmic reticulum, so a protein called the invariant chain (" Ii ") temporarily occupies the groove.
- The two chains of the class II molecule are inserted into the membrane of the endoplasmic reticulum.
- They bind (in their groove) one molecule of invariant chain.
- This trimolecular complex is transported through the Golgi apparatus and into vesicles called lysosomes.
Meanwhile foreign antigenic material is engulfed by endocytosis forming endosomes . These also fuse with lysosomes. Then,
- The antigen is digested into fragments.
- The invariant (Ii) chain is digested.
- This frees the groove for occupancy by the antigenic fragment.
- The vesicles move to the plasma membrane and the complex is displayed at the cell surface.
- a receptor ( TCR ) able to bind the peptide and flanking portions of the histocompatibility molecule (the hot dog in the bun) and
- CD4 molecules that bind the CD4 receptor (shown above as a yellow triangle) found on all class II histocompatibility molecules.
Interconnections Between the Class I and Class II Pathways
Cross-presentation: transferring exogenous antigens to the class i pathway.
Cross-presentation is the transferring of extracellular antigens like bacteria, some tumor antigens, and antigens in cells infected by viruses into the class I pathway for stimulation of CD8 + cytotoxic T cells (CTL). Only certain "professional" antigen-presenting cells (APCs) like dendritic cells can do this - use the class I as well as the class II pathways of antigen presentation.
Cross-presentation following infection by viruses is important because:
- Most viruses infect cells other than APCs (e.g., liver cells, epithelial cells of the lung) (and, of course, are intracellular in these).
- While viral antigens displayed on the surface of any infected cell can serve as targets for cytotoxic T cells (CTLs),
- the lack of any costimulatory molecules on the cell surface makes them poor stimulants for the development of clones of CTLs in the first place.
However, when an infected cell dies, it can be engulfed by a professional APC, and the antigens within it can enter the class I pathway. One mechanism:
- The dead cell is engulfed by endocytosis.
- The endosome that forms fuses with a lysosome and degradation of the dead cell begins.
- Antigens pass into the cytosol and are degraded in proteasomes.
- The peptides formed are then are picked up by TAP and inserted into class I MHC molecules and displayed at the cell surface — along with the costimulatory molecules needed to start a vigorous clonal expansion of CD8 + cytotoxic T cells.
Diverting Antigens from the Class I to the Class II Pathway
Autophagy provides a mechanism by which cells can transfer endogenous (intracellular) antigens into the class II pathway, for example
- self-proteins so as to be able to delete CD4 + T cells with receptors capable of attacking them and thus potentially capable of causing autoimmunity
- proteins synthesized by an infecting virus. In this way viral infection can generate CD4 + T cells as well as cytotoxic T cells (CD8 + )
B Lymphocytes: A Special Case
B lymphocytes are both antigen-receiving and antigen-presenting cells. They bind intact antigens (e.g., virus particles, proteins) with their B cell receptor (BCR). They can come in contact with these antigens by encountering them in the surrounding lymph or by being presented them by macrophages or dendritic cells. B lymphocytes process antigen by the class II pathway for presentation to T cells.
The process:
- B cells engulf antigen by receptor-mediated endocytosis
- The B cell receptors for antigen ( BCR s) are antibodies anchored in the plasma membrane.
- The affinity of these for an epitope on an antigen may be so high that the B cell can bind and internalize the antigen when it is present in body fluids in concentrations thousands of times smaller than a macrophage would need.
- The remaining steps of antigen processing occur by the same class II pathway described above for macrophages producing
- fragments of antigen displayed at the cell surface nestled in the groove of class II histocompatibility molecules.
- A CD4 + T cell that recognizes the displayed antigen is stimulated to release lymphokines.
- These, in turn, stimulate the B cell to enter the cell cycle.
- Because of the part they play in stimulating B cells, these CD4 + T cells are called Helper T cells (" Th ").
- The B cell grows into a clone of cells (called plasma cells )
- These synthesize receptors ( BCR s) with the identical binding site for the epitope but without the transmembrane tail.
- These antibodies are secreted into the surroundings.
Lipid and Polysaccharide Antigens
Lipid antigens.
- Lipid antigens are presented to T cells by cell-surface molecules designated CD1 ("cluster of differentiation" 1).
- Antigen-presenting cells express several different forms of CD1 at their surface. Each is probably specialized to bind a particular type of lipid antigen (e.g. lipopeptide vs glycolipid).
- The exposed surface of CD1 molecules forms an antigen-binding groove much like that of MHC molecules except that
- the amino acids in the groove are more hydrophobic than those in MHC molecules.
- Like protein antigens, lipid antigens are also presented as fragments, i.e., as a "hot dog in a bun".
Polysaccharide Antigens
Some bacterial polysaccharides ingested by APCs
- can be degraded in their lysosomes
- and presented to T cells by MHC class II molecules.
The binding of a T cell to an antigen-presenting cell (APC) is by itself not enough to activate the T cell and turn it into an effector cell: one able to, for examples,
- kill the APC (CD8 + cytotoxic T lymphocytes [CTLs])
- carry out cell-mediated immune reactions (CD4 + Th1 cells)
- provide help to B cells (CD4 + Th2 cells)
In order to become activated, the T cell must not only bind to the epitope (MHC-peptide) with its TCR but also receive a second signal from the APC. The receipt of this second signal is called costimulation . Among the most important of these costimulators are molecules on the APC designated B7 and their ligand on the T cell designated CD28 . The binding of CD28 to B7 provides the second signal needed to activate the T cell.
Although T cells may encounter self antigens in body tissues, they will not respond unless they receive a second signal. In fact, binding of their TCR ("signal one") without "signal two" causes them to self-destruct by apoptosis. Most of the time, the cells presenting the body's own antigens either
- fail to provide signal two or
- transmit an as-yet-unidentified second signal that turns the T cell into a regulatory T cell ( Treg ) that suppresses immune responses.
In either case, self-tolerance results.
REVIEW article
Predicting antigen presentation—what could we learn from a million peptides.

- 1 Department of Oncology, Ludwig Institute for Cancer Research, University of Lausanne, Lausanne, Switzerland
- 2 Swiss Institute of Bioinformatics (SIB), Lausanne, Switzerland
- 3 Department of Oncology, Ludwig Institute for Cancer Research, University Hospital of Lausanne, Lausanne, Switzerland
Antigen presentation lies at the heart of immune recognition of infected or malignant cells. For this reason, important efforts have been made to predict which peptides are more likely to bind and be presented by the human leukocyte antigen (HLA) complex at the surface of cells. These predictions have become even more important with the advent of next-generation sequencing technologies that enable researchers and clinicians to rapidly determine the sequences of pathogens (and their multiple variants) or identify non-synonymous genetic alterations in cancer cells. Here, we review recent advances in predicting HLA binding and antigen presentation in human cells. We argue that the very large amount of high-quality mass spectrometry data of eluted (mainly self) HLA ligands generated in the last few years provides unprecedented opportunities to improve our ability to predict antigen presentation and learn new properties of HLA molecules, as demonstrated in many recent studies of naturally presented HLA-I ligands. Although major challenges still lie on the road toward the ultimate goal of predicting immunogenicity, these experimental and computational developments will facilitate screening of putative epitopes, which may eventually help decipher the rules governing T cell recognition.
Introduction
Recognition of infected or malignant cells by T cells relies on the presentation of immunogenic self and non-self peptides at the cell surface. Two main pathways have been identified for antigen presentation and processing ( 1 – 3 ).
In the class I pathway, intracellular proteins are degraded into small peptides by the proteasome. These peptides are transported into the endoplasmic reticulum by the transporter associated with antigen processing (TAP) protein complex. There, they can bind to human leukocyte antigen class I (HLA-I) molecules in complex with beta2-microglobulin (β2m). After trafficking to the cell surface, the complexes may be recognized by CD8 T cells. HLA-I proteins are primarily encoded by three genes (HLA-A, HLA-B, and HLA-C), which are widely expressed in most cell types in human. In addition, specialized cell types can express HLA-E, HLA-F, or HLA-G genes. HLA-A, -B, and -C genes (hereafter referred to as HLA-I) are the most polymorphic genes in the human genome and over 12,000 distinct alleles are documented in the human population ( 4 ). Humans have in general different combinations of HLA-I alleles and, therefore, express up to six different HLA-I proteins (two for each gene). HLA-I molecules bind short peptides, mainly 9–11 amino acids, and different HLA-I alleles have distinct binding specificities, which implies that a broad spectrum of peptides can be displayed across different individuals.
In the class II pathway, peptides coming from the degradation of phagocytosed extracellular proteins are presented on HLA-II molecules for recognition by CD4 T cells ( 5 ). In addition, endogenous proteins can be presented on HLA-II molecules when degraded through autophagy ( 6 ). HLA-II proteins are encoded by several genes (HLA-DRA, HLA-DRB1,3,4,5, HLA-DPA1, HLA-DPB1, HLA-DQA1, HLA-DQB1) and also show a very high level of polymorphism in the humans (except for HLA-DRA). HLA-II form heterodimers (HLA-DRA/HLA-DRB1,3,4,5; HLA-DPA1/HLA-DPB1 and HLA-DQA1/HLA-DQB1). These dimers bind longer peptides (12–20 amino acids) within an open-ended peptide-binding site. Several other steps are involved in presentation of class II epitopes, such as loading on HLA-II molecules catalyzed by HLA-DM, peptide exchange catalyzed by HLA-DO, the presence of other enzymes such as cathepsins or pH gradients ( 7 – 10 ). Unlike HLA-I, HLA-II molecules are mainly expressed on specific professional antigen-presenting cells (pAPCs) such as dendritic cells or B cells ( 1 ), and rarely also by cancer cells such as melanoma ( 11 ). pAPCs can also uptake exogenous antigens and present them on HLA-I ( 12 ). This process is called cross-presentation, and it is crucial for priming of naïve T cells ( 13 , 14 ). Altogether, the cellular antigen processing and presentation machinery ensures that the restrictive loading of either intracellular (class I) or extracellular (class II) peptides of the right length will take place in specialized cellular compartments.
The set of peptides presented on HLA molecules is called the HLA peptidome, also referred to as immunopeptidome or HLA ligandome. The HLA peptidome is a rich and complex repertoire of peptides that inform T cells about abnormalities in the genome, transcriptome, and proteome of infected or malignant cells ( 15 – 17 ). It is constantly modulated by HLA or peptides’ source protein expression levels, by posttranslational modifications and by the many enzymes, chaperones, and transporters that comprise the antigen processing and presentation machinery ( 7 , 18 – 20 ). In particular, the catalytic subunits of the constitutive proteasome, the immunoproteasome, and the thymic proteasome are tightly regulated, leading to the production of distinct repertoires of presented peptides in different cell types and under different conditions ( 21 – 24 ).
Historically, the study and predictions of class I and class II T cell epitopes have mainly developed in the field of infectious diseases, and large datasets of peptides displayed at the surface of infected cells and recognized by T cells are available from HIV, dengue, or influenza ( 25 , 26 ). In the field of cancer immunology, tumor-associated antigens (defined here as genes expressed in cancer cells and not, or very poorly, in normal cells) have received much attention for almost 30 years ( 27 ). For instance, T cell recognizing specific epitopes of NY-ESO or MAGE-1 proteins can be found in melanoma patients, indicating that the immune system can mount a response against tumor-specific antigens ( 27 – 29 ). More recently, many evidences have been accumulated indicating that cancer cells express unique mutated antigens, the so-called neoantigens, which can be recognized by the patients’ own (autologous) T cells ( 15 , 30 – 35 ). The total number of somatic mutations in some tumors has been shown to correlate with the therapeutic efficacy of checkpoint blockade antibodies ( 36 – 39 ), suggesting that neoantigens could play an important role in tumor immune recognition. Moreover, several studies demonstrated clinical benefit mediated by the administration of highly enriched populations of neoantigen-reactive CD4 + and CD8 + T cells ( 34 , 40 ) and by neoantigen-based vaccines ( 41 , 42 ). Potential neoantigens are typically predicted first by identifying non-synonymous alterations from next generation sequencing data and second by predicting the binding to HLA molecules of peptides encompassing these non-synonymous genetic alterations ( 43 ). For these reasons, predictions of peptides presented on HLA-I and HLA-II molecules have gained renewed interest in the field of tumor immunology. Predicted neoantigens need to be then experimentally validated for HLA binding and immune recognition in vitro ( 44 – 47 ).
Here, we review approaches developed for predicting antigen presentation in human cells, with a focus on the latest experimental and computational developments to take advantage of in-depth and accurate mass spectrometry (MS) data of HLA peptidomics. Our aim is to describe the main steps of antigen presentation that proved to be successful in making quantitative predictions of antigens. The more biological aspects of antigen presentation and processing are covered in many other reviews ( 1 – 3 , 8 ).
Main Sources of HLA Ligand Data
A cornerstone in our ability to understand and predict antigen presentation has been the experimental identification of specific peptides interacting with HLA molecules. First, from an experimental point of view, HLA-I molecules do not fold stably in the absence of a ligand and, therefore, all biochemical, structural, and functional studies of HLA-I molecules rely on the availability of known HLA-I ligands. Second, all computational methods to predict HLA ligands at a large-scale use data-driven approaches based on sequence patterns identified within known ligands.
Two main classes of experimental assays have been developed to identify HLA ligands. The first class of assays consists of in vitro assays. For HLA-I molecules, refolding assays use conformational pan HLA-I antibodies to test whether the HLA-I complex is properly folded in the presence of a peptide ( 48 – 52 ). Peptide-rescuing assays consist of a photo-cleavable peptide that is stripped by UV radiation in the presence of another peptide ( 53 – 55 ). Competitive assays with radiolabeled peptides have been used to determine relative affinity (IC50) ( 56 ). Dissociation assays based on radiolabeled β2m have been used to probe the stability of peptide–HLA-I complexes ( 57 , 58 ). Surface plasmon resonance techniques can be used to measure actual Kd values ( 59 ). In vitro binding assays have also been used for HLA-II ligands ( 60 – 62 ). Compared to class I ligands, screening of class II ligands at high throughput is facilitated since HLA-II molecules have an open-ended peptide-binding site. Therefore, peptides can be fixed on plates, which allow for the use of peptide microarrays ( 63 ), or directly encoded in different display systems such as phage or yeast display ( 64 , 65 ).
In vitro binding assays play a central role in our ability to identify T cell epitopes from viral or cancer-specific antigens ( 66 , 67 ). When used in combination with state-of-the art predictions tools, they enable rapid validation of predicted targets and are currently key to most neoantigen discovery approaches in cancer immunotherapy ( 30 , 31 , 68 , 69 ). The main caveat of in vitro assays for HLA-I ligands is that the peptides have to be determined a priori and chemically synthesized, since both the C- and N-terminus of most HLA-I ligands need to be free in most cases. This limits the use of high-throughput and unbiased peptide screening technologies. Furthermore, the involvement of the components of the antigen-loading complex is missing in in vitro binding assays and, therefore, signals related to antigen loading in vivo cannot be captured.
The second type of experimental assays for HLA ligand identification is based on MS measurement of eluted HLA-binding peptides. This approach is the only methodology to comprehensively interrogate the repertoire of HLA ligands presented naturally in vivo ( 16 , 18 , 70 , 71 ). The best-established HLA peptidomics methodology is based on immunoaffinity purification (IP) of HLA complexes from detergent solubilized lysates, followed by extraction and purification of the peptides. Typically, either anti-pan-HLA class I, anti-HLA-DR, or anti-pan-HLA class II monoclonal antibodies are used. The extracted peptides are then separated by high-pressure liquid chromatography and directly injected into a mass spectrometer. The resulting spectra obtained from the fragmentation of the peptides are compared with in silico generated spectra of peptides from protein sequence databases with MS search tools. Therefore, this search is limited to the available databases, usually the annotated human proteome. Moreover, peptides that have features that make them incompatible with ionization, those that are too hydrophobic or too hydrophilic, might not be detected with standards methods. With the new generation of mass spectrometers, thousands of HLA ligands can be identified per sample ( 15 , 18 , 72 , 73 ). Cell lines, including human cancer cell lines, tumors, healthy tissues, and body fluids such as plasma have been subjected to HLA peptidomics analyses ( 18 , 70 – 84 ). However, MS-based HLA peptidomics approaches have limited sensitivity and require a relatively large amount of biological sample (~1 cm 3 of tissue or 1 × 10 8 cells) ( 21 ). Furthermore, despite major improvement in the quality of HLA peptidomics data, one can never exclude small residual contaminations from co-eluted peptides or wrong annotation of spectra depending on the false discovery rate threshold used in spectral searches.
Dedicated proteogenomics computational pipelines for customized reference databases have been developed to expand the search space beyond the canonical human proteome. Customizing references to include somatic alterations observed in tumors have been used for direct identification of neoantigens by MS in murine and human cancer cell line models ( 31 , 35 , 80 , 85 ), B cell lymphomas ( 86 ), and melanoma tissues ( 15 ). Similar approaches were also used for other cryptic peptides resulting from unconventional coding sequences in the genome ( 87 ) and new open reading frames ( 88 ) (see Non-Canonical HLA-I Ligands).
Historically, the first HLA-I motifs (e.g., HLA-A02:01) were found by looking at peptide sequences of eluted ligands identified by MS ( 89 , 90 ). To overcome the fact that eluted peptides come from up to six HLA-I alleles in unmodified cell lines or tissue samples, two experimental approaches have been developed. The first approach consists of transfecting a soluble HLA allele into a cell line and pulling down only the soluble HLA-I molecules in complex with their ligands ( 91 , 92 ). While it has been shown that the repertoire of peptides presented on transfected soluble HLA-I and the endogenous membranal HLA-I molecules are highly similar ( 93 ), the non-physiological expression level of the soluble HLA-I molecules and the potential different environment in the loading compartment could affect the overall peptide repertoire. Furthermore, endogenous HLA-I alleles can be shaded or naturally secreted from cells in culture ( 94 ) and could contaminate the secreted peptidome ( 75 ). Nevertheless, this approach proved very powerful to identify HLA-I motifs ( 77 , 78 , 95 – 97 ). Of particular interest is the study by Di Marco and co-authors where the motifs of 15 HLA-C alleles could be determined, together with motif for HLA-G01:01 ( 75 ). This detailed view of HLA-C alleles binding specificities enabled the authors of this study to identify for the first time specificity determinant residues in the HLA-C-binding site that provide likely molecular mechanisms explaining the differences observed between HLA-C binding motifs. The second experimental approach consists of using genetically modified cell lines that express only one allele ( 98 , 99 ) and was used to study binding motifs of highly similar alleles, like HLA-B27:02 to HLA-B27:09 ( 100 ). This approach was also recently used to screen 16 HLA-A and HLA-B alleles, and this work confirmed that predictors trained on MS data could improve predictions of naturally presented HLA-I ligands ( 70 ). One advantage of this approach is that theoretically all peptides come from one single allele (see above for potential sources of contaminations). In parallel, we and others introduced computational techniques based on motif deconvolution ( 72 , 101 ) and peptide clustering ( 102 , 103 ) to accurately determine HLA-I restriction of eluted ligands from pooled samples without requiring to experimentally isolate each HLA-I allele and without relying on HLA-I ligand predictors (see below for a detailed description of these approaches).
Comparison of MS and In Vitro Data
Until 2012, the number of MS datasets was significantly lower than in vitro data (Figure 1 ), which partly explains why in vitro binding data were mainly used for training HLA-I ligand predictors. However, the situation has changed quite dramatically over the last 4 years. Combining data from IEDB ( 25 ) together with recent HLA peptidomics studies (see Supplementary Material), we can observe that roughly 10 times more unique HLA-I ligands and three times more unique HLA-I–peptide interactions are currently available from MS studies (Figure 1 , the lower number of interactions than peptides for MS data comes from the fact that several MS samples did not have HLA typing information or allele restriction could not be determined with motif deconvolution). The coverage of HLA-I alleles is also larger in HLA peptidomics samples compared to in vitro binding data (Figure 1 ). Moreover, all curves for MS data do not show signs of saturation, suggesting that these numbers are likely to further increase in the coming years, especially with the growing interest in HLA peptidomics profiling of cancer samples from patients with diverse ethnic backgrounds for neoantigen discovery ( 15 ). Similar observations hold for HLA-II ligands, where the number of unique peptides identified by MS largely exceeds the number of peptides identified in in vitro assays. However, the number of HLA-II alleles with documented ligands is still larger for in vitro binding data. This likely reflects the fact that HLA-II ligands are easier to screen in a high-throughput way using peptide microarrays, and that allele restriction in HLA-II peptidomics data is still more difficult to determine with motif deconvolution or peptide clustering than for HLA-I peptidomics data.

Figure 1 . Analysis of HLA-I and HLA-II ligands obtained from human leukocyte antigen (HLA) peptidomics studies and in vitro assays. The number of unique HLA-I ligands, the number of unique interactions, the number of HLA alleles with at least one ligand, and the number of HLA alleles with at least 100 ligands are displayed for both class I and class II, as a function of years (cumulative distributions).
Modeling HLA-I Binding Specificity
Allele-specific predictors.
Modeling HLA-I-binding specificity has been carried out for almost 30 years since the first evidence of HLA-I motifs. Early studies used simple sequence motifs [e.g., xLxxxxxx(L/V) for HLA-A02:01]. However, as more data started to accumulate, it became clear that simple motifs were too restrictive and not quantitative enough. To overcome these limitations, position weight matrices (PWM) (equally referred to as Position Specific Scoring Matrices or simply scoring matrices) were introduced ( 104 – 107 ). The basic idea is to compute the frequency of each amino acid at each position in a set of (pre-aligned) peptides. The score of a new peptide can then be computed by multiplying the PWM entries corresponding to the sequence of the new peptide (see Supplementary Material). Although the idea of computing amino acid frequencies is relatively simple to understand, several steps are important when building a predictor based on PWMs. First, one has to consider the amino acid background distribution and use this distribution to renormalize the scores (see Supplementary Material). In most existing approaches, amino acid frequencies of the human proteome have been used. However, this approach may not be fully justified when using viral epitopes to train predictors. Similarly, eluted HLA-I ligands do not show the same amino acid distribution as human proteins and much lower frequency of cysteine has been reported by ourselves and others ( 70 , 72 ). As such, the optimal choice of background distribution may depend on the origin (both biological and technical) of the data. Second, in most cases, estimating the frequency of amino acids occurring only a few times (or never) at a given position is highly susceptible to statistical noise. To address this issue, pseudo-counts are often used. A widely used approach is based on the BLOSUM62 matrix (see Supplementary Material) ( 105 , 108 , 109 ). Third, biases due to the design of specific experiments can be found in many in vitro datasets. For instance, if a mutagenesis was carried out at a fairly non-specific position in a given epitope, many sequences will have identical amino acids at all positions except the one used in the mutagenesis. One way to correct for such biases is to add a weight to all peptides that is inversely proportional to the number of highly similar sequences in the dataset (see Supplementary Material).
Since the last decade, most allele-specific HLA-I ligand predictors use machine learning frameworks such as neural networks, hidden Markov Models, support vector machines, or convolutional neural networks ( 110 – 114 ). One attractive aspect of these models is the ability to consider potential correlations between different positions within HLA-I ligands. For instance, we recently observed in HLA-B07:02 ligands that arginine is preferred at P3 or at P6, but not at both positions at the same time ( 101 ). This type of correlation is not captured by simple PWMs. However, it is still unclear how frequent these correlations are for HLA-I ligands. In particular, although many studies reported improved predictions of HLA-I ligands using machine learning algorithms ( 112 , 115 ), one has to be careful before concluding that correlation patterns are prevalent, since improvement in prediction accuracy may also result from more robust regularization frameworks. Finally, machine learning approaches are also susceptible to overfitting and correcting for potential biases in training sets can be more challenging than with simple PWMs.
Pan-Allele Predictors
Enough experimental ligands are available for roughly 100 HLA-I alleles, which represents only a small fraction of the >12,000 HLA-I alleles observed in the human population. To address this issue, pan-allele predictors have been introduced, where the input of the algorithm consists of both the sequence of the ligand and the sequence of the HLA-I allele (or of its binding site) ( 107 , 116 – 118 ). These algorithms are powerful at capturing correlations between amino acids in the HLA-I-binding site and in the ligand. The most widely used and likely the most elaborate pan-specific algorithm is the NetMHCpan tool ( 117 ), which includes several features specific for HLA-I molecules, such as combining peptides of different lengths in the training and incorporating peptide length preferences.
Table 1 summarizes some of the most common predictors, together with information about the algorithm that is used, the type of training data and the output.

Table 1 . Summary of some of the most recent or most widely used human leukocyte antigen (HLA)-I predictors with available web interface or code repository.
Choosing the Right Training Set
While extensive work has been performed to optimize the algorithms used in HLA-I predictors, less attention has been devoted to the choice of the training set. Prior to 2016, most approaches aimed at predicting binding affinity values (i.e., IC50) and, therefore, were trained on in vitro data mainly obtained from IEDB ( 25 ). Although high accuracy could be reached for many common alleles, several potential biases suggest that such data can be suboptimal for training predictors. In particular, it is important to remember that most HLA-I ligands tested in vitro for binding were first predicted with older versions of HLA-I ligand predictors [some exceptions that used random peptide libraries include Ref. ( 58 )]. Unfortunately, this can induce circularity when using these data to retrain predictors, and such biases are difficult to detect and correct for. Of note, the same circularity issue can also affect several published MS datasets when HLA-I ligand predictors or motifs were used to assign allele restriction and filter noise. Here, we argue that high-quality MS data not filtered with existing predictors provide a powerful solution toward overcoming the potential circularity inherent to many in vitro binding data.
Using MS Data for Identifying HLA-I Motifs and Training Predictors
Mono-allelic samples or transfected soluble HLA-I alleles have been used since many years to study the binding motifs of specific HLA-I molecules ( 91 , 92 ). However, due the experimental work implied by such approaches, they were never applied to a large panel of HLA-I alleles [the largest studies consist of 16 alleles for mono-allelic cell lines ( 70 ) and 17 alleles for transfected soluble HLA-I alleles ( 75 )]. For pooled HLA peptidomics dataset, the impossibility to experimentally assign allelic restriction was often considered as an important hurdle to use such data toward studying HLA-I-binding motifs.
However, in the last few years, it became clear that pooled HLA peptidomics data can be used to study HLA-I motifs and improve predictions, thereby overcoming the need of genetically modifying cell lines or transfecting soluble HLA-I alleles. The first attempt to determine HLA-I-binding motifs from pooled HLA peptidomics data was published in 2015 ( 18 ). A year later, we published the first evidence that such data can be used to improve predictions of HLA-I ligands ( 101 ). Since then, many studies have confirmed these results both for the identification of new motifs ( 72 , 81 , 102 , 103 , 119 ) and for improving predictions of HLA-I ligands by integrating MS data in the training of predictors ( 70 , 72 , 117 , 120 ).
As of today, two algorithms have been used for motif deconvolution and peptide clustering of pooled HLA peptidomics data. One of them (MixMHCp) is based on mixture models and was initially developed for multiple specificity analysis in large PDZ or SH3 ligand datasets obtained by phage display ( 121 – 123 ). In this framework, the idea is to let the algorithm infer K distinct PWMs that optimally model the eluted peptides ( 101 ). Since peptides identified by MS come from K different HLA-I alleles ( K ≤ 6), it is not surprising that the motifs that optimally describe the data correspond precisely to the specificity of these alleles. The other algorithm (GibbsCluster) is based on simulated annealing to group the peptides into different clusters optimizing a global cost function that models how well each peptide fits into its respective cluster ( 103 , 124 ). Somehow unexpectedly, both algorithms were initially developed for other purposes (i.e., multiple specificity analysis for MixMHCp and simultaneous clustering and alignments of short peptides for GibbsCluster) and their use for motif identification in HLA peptidomics data was realized only later ( 18 , 101 , 102 ). The two approaches have many conceptual similarities since the likelihood function optimized in MixMHCp differs only slightly from the cost function optimized in GibbsCluster. In practice, the two algorithms lead most of the time to very similar results for HLA-I peptidomics data ( 101 ) and nearly identical motifs as those obtained from mono-allelic samples or transfected soluble alleles ( 72 ) (see also examples in Figure S1 in Supplementary Material). In some cases, as we have reported, the mixture model tends to be slightly more sensitive to identify motifs supported by few peptides, such as those describing HLA-C alleles ( 101 ). Conversely, the GibbsCluster has several advantages, such as the ability to combine peptides of different lengths and the simultaneous clustering and alignment of the peptides (which is critical for HLA-II ligands) ( 102 , 103 ). Both methods can be used as command line or through webservers (see http://www.mixmhcp.org and http://www.cbs.dtu.dk/services/GibbsCluster-2.0/ ). The availability of these algorithms strongly supports the notion that allele assignment in MS data should not be done based on HLA-I ligand predictors, since this may remove all peptides that are not well modeled with existing predictors, and hence bias determination of motifs and prevent improving the predictors. It is also important to emphasize that accurate motif deconvolution requires a large number of peptides, and ideally, many samples to test the robustness of the motifs ( 72 ). For this reason, it is likely the combination of higher accuracy and throughput of MS instruments ( 18 ) together with these novel algorithms that enabled accurate HLA-I motifs identification in pooled HLA peptidomics data.
Annotation of the motifs deconvolved from pooled HLA peptidomics data can be done in different ways. For alleles for which a reasonable description of the motifs is known, one can simply compare the motifs found in MS data to the known references ( 18 ). Using Euclidean distance to quantify the similarity between the PWMs appears to provide stable results and most of the time the mapping is quite obvious ( 72 , 101 ). If the motifs are not known, two approaches have been developed. One fully unsupervised approach was proposed by ourselves based on cooccurrence of HLA-I alleles across different samples ( 72 ). In this way, we could identify and annotate HLA-I motifs for more than 40 alleles, including 7 alleles that had no experimental ligands at the time of this study. Another semi-supervised approach that works well in most cases consists of comparing with motifs predicted from pan-allele predictors such as NetMHCpan ( 119 ).
An important limitation of motif deconvolution approaches comes from the fact that motifs for some alleles (especially HLA-C alleles) are more difficult to detect in many samples. Also, in the presence of highly similar motifs (e.g., HLA-A23:01 and HLA-A24:02, or HLA-C07:01 and HLA-C07:02), the two motifs often cannot be split ( 72 ). Because of this, not all HLA peptidomics datasets are appropriate for training predictors for each allele expressed in the corresponding sample. This limitation can be alleviated by considering large collections of HLA peptidomics studies and focusing on cases where the motifs are clearly visible and can be unambiguously annotated ( 72 ). Finally, it is sometimes useful to consider more motifs than the number of alleles in order to identify motifs for each allele (Figure S2 in Supplementary Material).
Figures 2 – 4 summarize the HLA-A, HLA-B, and HLA-C motifs currently available by combining motifs deconvolved from recent MS studies together with IEDB data (see Supplementary Material). As expected, the clustering based on the similarity between the motifs (see Supplementary Material) broadly recapitulates the supertype assignment for HLA-A and HLA-B alleles and helps highlighting differences among alleles classified within the same supertypes.

Figure 2 . Hierarchical clustering of HLA-A alleles based on their binding specificity. Stars indicate cases where only in vitro binding data were available to generate the motifs. In all other cases, only mass spectrometry data were used. Name colors and their descriptions in the legend indicate supertypes as defined in Ref. ( 185 ).

Figure 3 . Hierarchical clustering of HLA-B alleles based on their binding specificity. Stars indicate cases where only in vitro binding data were available to generate the motifs. In all other cases, only mass spectrometry data were used. Name colors and their description in the legend indicate supertypes as defined in Ref. ( 185 ).

Figure 4 . Hierarchical clustering of HLA-C alleles based on their binding specificity. Only mass spectrometry data were used. Peptides from both single allele and deconvolved pooled HLA peptidomics samples were used (see Supplementary Material).
Biases in MS Data
While MS data are not suffering from the potential circularity present in many in vitro binding data, they are not free from any biases. First, as already mentioned, only peptides that are part of the database used for spectral searches can be detected in HLA peptidomics data, or else, the less accurate de novo method may be applied. This has direct implication for cysteine-containing peptides. Since this amino acid can be chemically modified by oxidation and as such modifications are typically not included in standard MS searches, cysteine occurs at very low frequency in HLA peptidomics datasets. Attempts to correct for this bias when training predictors tried to renormalize PWMs based on observed amino acid frequencies at non-anchor positions ( 72 ) or expand the MS spectral search to include modified cysteines ( 70 ). Second, peptides that are too hydrophobic or too hydrophilic might be missed applying the common purification methods that rely on retaining peptides through hydrophobic interactions with the solid phase. Furthermore, some peptides have features that make them incompatible with ionization or lead to poor fragmentation. Combining fragmentation methods, such as higher-energy collision-induced dissociation and electron-transfer dissociation, have been shown to improve spectra annotation of HLA peptides ( 73 ). Despite these limitations, inspection of HLA peptidomics data and comparison with motifs obtained from in vitro data did not reveal major differences, except for the low frequency of cysteine [slightly higher frequency of charged amino acids at some positions has been reported in some studies ( 101 , 102 )]. Third, immuno-purification based MS data cannot distinguish between HLA-I ligands presented on the cell surface from those resident in the ER. This can be achieved by purifying HLA-I peptides from the cell surface by mild acid elution ( 125 , 126 ). However, in a head-to-head comparison, the IP method outperformed the mild acid elution in terms of peptide recovery ( 127 ). Last, when considering MS data, it is important to remember that these peptides come from human proteins and that proteins or domains within proteins can display significant homology (especially for class II ligands where in addition many peptides can originate from the same core region). This can artificially enhance the frequency of some amino acids. This issue is especially important when building random models of MS data to infer whether amino acid frequencies (either within a motif or at flanking regions) differ from what is expected by chance.
Modeling HLA-II-Binding Specificity
Predictions of HLA-I ligands, especially with the recent incorporation of high-quality MS data in the training of predictors, have reached a high level of accuracy ( 70 , 72 , 117 , 120 ). The situation is unfortunately not the same for HLA-II ligands, which are still much more difficult to predict despite the large amount of experimental data acquired over the years (Figure 1 ). Several challenges arise when modeling the binding specificity of HLA-II alleles. First, HLA-II alleles tend to have more degenerate and less specific motifs. Second, all current approaches rely on first aligning peptides with tools such as NN-align ( 128 ). Although these tools have been optimized to handle HLA-II ligands, automated alignment of small peptides is known to be a difficult computational problem. Finally, the fact that HLA-II molecules form dimers further increases the diversity for HLA-DP and HLA-DQ alleles where both members of the dimers are polymorphic. Allele-specific HLA-II ligand predictors include NetMHCII ( 129 ), ProPred Singh ( 130 ), MHCPred ( 131 ), TEPITOPE ( 132 ), and consensus methods ( 133 ). Pan-specific class II predictors mainly consist of NetMHCIIpan ( 129 ). While all these predictors show better than random performances, their accuracy is lower than for HLA-I ligand predictors. This may be due to the challenges of determining class II motifs, as well as to the complex machinery of class II presentation, whose specificity is still poorly understood from a quantitative and predictive point of view [see Ref. ( 7 – 10 ) for a detailed discussion of the more biological aspects of this process and the importance of HLA-DM and other enzymes]. In particular, it appears that properties such as conformational flexibility play a role in loading onto HLA-II molecules ( 134 ), and these properties are difficult to predict directly from peptide sequences.
Whether similar improvement for class II predictions as for class I will be reached by incorporating class II peptidome data in the training of algorithms has not been investigated at a large scale. Nevertheless, it has been recognized already long ago that eluted ligands could provide important information about HLA-II-binding motifs ( 135 ). More recently, HLA-II peptidomics was performed in BALB/c and C57BL/6 mice and demonstrated that clear motifs for H-2 I-Ad and H-2 I-Ab could be obtained ( 136 ). A subsequent study suggested that predictors trained on these data perform better than NetMHCIIpan when repredicting the MS data ( 137 ). A similar strategy was carried out in transgenic DR1+ and DR15+ mice to identify the motifs of these two alleles ( 138 ). Recent studies also indicate that motif deconvolution with the GibbsCluster algorithm may work in pooled HLA-II peptidome datasets ( 21 , 139 ), which could lead to refinement of HLA-II motifs and improved predictions in the coming years, as suggested in a recent preprint ( 140 ). However, the results are still more challenging to interpret and some motifs predicted by GibbsCluster are difficult to annotate, while the motifs for some alleles are sometimes not detected ( 21 , 139 , 141 ).
Investigating Other Properties of HLA–Peptide Interactions
Many other important properties of HLA-I molecules beyond the 9-mer-binding motifs themselves can be studied through the analysis of HLA peptidomics data.
Peptide Length Distribution
Arguably, the most important information beyond the binding motifs that can be extracted from MS data is the characterization of peptide length distributions. Many studies have demonstrated high heterogeneity of peptide length distributions between different alleles, with alleles such as HLA-B51:01 displaying high frequency of 8-mers (only slightly smaller than 9-mers) and very few longer peptide, while others such as HLA-A01:01 show high frequency of longer (≥12 amino acids) peptides, which can still be recognized by T cells ( 15 , 70 , 97 , 103 , 142 ). Structurally, most longer peptides are known to form bulges, with anchor residues conserved at the second and last positions of the peptides. Some patterns emerged from analysis of peptide length distributions. For instance, HLA-I alleles with anchor residues at middle positions (e.g., HLA-B08:01, HLA-B14:01, HLA-B14:02, HLA-B37:01) displayed peptide length distributions peaked at 9-mers, which is consistent with the fact that the middle anchor residue needs to be structurally conserved in the presence of an anchor at such positions ( 101 ). The study by Trolle and co-authors ( 97 ) demonstrated that peptide length distributions observed in MS data for five alleles could not be simply explained by differences in binding affinity, suggesting that the pool of peptides available for loading in the ER is skewed toward 9-mers. This likely implies that predictors trained on MS data will differ from those predicting binding affinity when comparing peptides of different lengths. In a recent preprint ( 143 ), we performed a large-scale analysis of peptide length distributions across 85 HLA-I alleles and could identify clusters of HLA-I molecules based on the similarity of their peptide length distributions. Peptide length distribution has been incorporated into the latest versions of NetMHC and NetMHCpan, by adding one additional input node encoding for peptide length in the neural networks ( 110 , 117 ), and into MixMHCpred by directly fitting distributions observed in MS data ( 143 ).
As observed in our recent paper ( 21 ), peptide length distribution can also be affected by different treatments such as INFγ likely due to modulating the activity of catalytic subunits of the proteasome, and these aspects are not captured by existing predictors.
C- And N-Terminal Extensions
Human leukocyte antigen peptidomics data have been instrumental in exploring non-canonical binding modes in HLA-I ligands. In particular, several recent studies have used MS data to study C- and N-terminal extensions in HLA-I ligands. Although such extensions had been identified long ago [first crystal structure in 1994 ( 144 ), PDB:2CLR, followed by another structure in 2009 ( 145 ), PDB:3GIV], their prevalence had remained unclear. In 2016, HLA peptidomics profiling and X-ray crystallography were combined to explore C-terminal extensions in HLA-A02:01 and demonstrated that such extensions were especially common among peptides originating from pathogens ( 146 ). This was followed by additional work that better described the structural mechanisms and cellular origin of such extensions ( 147 ). N-terminal extensions have been identified in HLA-B57:01 ( 148 ) and HLA-B58:01 ( 149 ). More recently, we have demonstrated that C-terminal extension occur in a substantial fraction of HLA-I molecules and can be recognized by CD8 T cells ( 120 ). Our work further enabled us to identify both sequence and structural features predictive of such extensions. In particular, it appeared that C-terminal extensions are especially frequent in alleles displaying specificity for positively charged residues at the last anchor position (e.g., HLA-A03:01, HLA-A31:01, HLA-A68:01). While MS data potentially provide a rich source of information about C- and N-terminal extensions, identifying these extensions by looking at the sequence of the peptides can be challenging, especially when the residue at the extension has similar specificity as the anchor residue (i.e., same residues at P9 and P10 for putative C-terminal extensions, same residues a P2 and P3 for putative N-terminal extensions). Our work suggests that many ambiguous cases may actually follow the bulging conformation ( 120 ).
Posttranslationally Modified HLA-I Ligands
Posttranslationally modified peptides have been identified by MS analysis of eluted ligands ( 15 , 150 – 152 ). These include mainly phosphorylated peptides, which can be recognized by T cells ( 153 – 155 ). Phosphorylation was observed to occur mainly at position 4 ( 15 ), suggesting that it does not impact too much the binding to the HLA-I molecules. Existing HLA-I ligand predictors do not include phosphorylated peptides, although the increasingly larger MS datasets of phosphorylated HLA-I ligands suggest that predictions of phosphorylated HLAI ligands may soon become feasible. As for now, one approach is to treat the phosphorylated residue as its unmodified counterpart and use available predictors to predict such ligands.
HLA-II Molecules
Fewer studies used MS data to investigate properties of HLA-II molecules other than the actual-binding motifs. Studies reported broad peptide length distributions peaked around 15-mers ( 15 , 21 , 139 , 156 , 157 ), but it is still unclear to what extent distinct alleles show distinct peptide length distributions. Other properties of HLA-II molecules that could be studied based on MS data include the cellular origin of class II peptides ( 156 , 158 , 159 ) and the impact of different biological processes such as autophagy ( 160 ). MS studies also indicated preference for proline at the second and second to last position of peptides degraded in the endolysosomal pathway ( 156 , 161 ), and preference for lysine at the C-terminus and for aspartate at the N-terminally flanking residue of class II epitopes degraded in the cytosolic pathway ( 156 ). Along these lines, many studies support the idea that presentation of class II peptides is not only driven by the binding specificity to the HLA-II molecules but also involves some (still uncharacterized) specificity in the processing machinery, flanking regions ( 162 ), or presentation hotspots in the human proteome ( 159 ).
Considering the increasingly higher quality and throughput of class II HLA peptidomics data ( 15 , 21 , 86 , 138 , 139 ), we anticipate that analysis of HLA-II peptidomes will further enable researchers to investigate new properties of HLA-II molecules. For instance, it will be interesting to see whether the presence of bulging class II ligands, as recently reported from an analysis of in vitro binding data ( 163 ), can be confirmed in large-scale unbiased MS data.
Antigen Presentation—Beyond Binding to HLA
Integrating cleavage site and tap transport predictions, signals from flanking regions and other proteomic information.
Mass spectrometry-based HLA peptidomics analysis can reveal crucial information about the rules underlying the biogenesis of the HLA peptidome, including signatures of cleavage site specificity, influence of source protein expression or other patterns characterizing naturally presented HLA ligands. Predictions of cleavage sites have been available since many years and have been used to narrow-down the list of predicted HLA-I ligands ( 164 ). Although some improvement has been observed, cleavage site predictions have only a limited effect on prediction accuracy of naturally presented HLA ligands. For this reason, it is not widely used in many existing pipelines for neoantigen predictions from exome sequencing data, for instance. Predictions of TAP transport has also been integrated with affinity and cleavage site predictions to model antigen presentation ( 165 – 167 ). Interrogation of properties of thousands of HLA-I ligands source-proteins has revealed that the proteome is not randomly sampled. Several biological determinants correlate with presentation, such as level of translation ( 71 ), expression, and turnover rate ( 18 ) and selective regions of the human proteome ( 71 ). Specific amino acid signals in flanking regions of naturally presented HLA-I ligands, like lower frequency of proline, have also been demonstrated ( 70 ). While binding to HLA still appears to be the most selective step of class I antigen presentation, integrating these additional features into a single predictor further improves the accuracy of predictions of naturally presented peptides ( 70 , 71 ).
Presentation Hotspots
After deep interrogation of HLA peptidomics large scale data, we and others have recently suggested that HLA ligands are not randomly distributed along the protein sequences but are located within “hotspots” ( 15 , 71 ), which fit proteasomal cleavage, peptide processing, and HLA-binding rules ( 168 ). Recently, we envisioned that these hotspots reflect regions of proteins with enhanced proteasomal or endosomal peptide production prior to HLA loading and may, therefore, provide complementary information to HLA-binding predictions ( 159 ). To this end, we collected a large dataset of MS detected HLA class I and class II ligands from different cancer and healthy tissues and variety of cell lines. We used this dataset to score potential neoantigens based on how well their un-mutated source proteins are naturally presented. In a proof of concept study, we tested this hypothesis with published data ( 33 ) and could show that MS-based features improved the prioritization of confirmed neoepitopes ( 159 ). Large scale databases of HLA peptidomics data capture the global nature of the in vivo peptidome averaged over many HLA alleles and, therefore, reflect the propensity of peptides to be presented, which can complement binding-affinity predictions.
Future Perspectives
Expanding the description of hla motifs.
Accurate and unbiased binding motifs are available for a bit more than 100 HLA-I alleles (Figures 1 – 4 ). This is only a tiny fraction of the >12,000 HLA-I alleles listed in IMGT/HLA database ( 4 ). For this reason, much has still to be learned about the specificity of HLA-I molecules. We anticipate that the ability to deconvolve HLA-I motifs from pooled HLA peptidomics data will play an important role to expand our understanding of HLA-I-binding specificities. This is especially promising in light of the current interest in using MS to identify neoantigens in cancer patients. However, even with the current efforts in HLA peptidomics, extrapolation of the curves in Figure 1 suggests that experimentally determined HLA-I ligands will remain available for only a small fractions of HLA-I alleles in the coming years. For this reason, development of pan-specific HLA-I ligand predictors leveraging high-quality MS data available for a few (~100) alleles to model the binding specificity of other alleles are expected to play an important role in broadening the scope of HLA-I ligand predictions to rarer alleles without document ligands ( 117 ). Accurate and in-depth HLA peptidomics data will also likely play an important role in improving our understanding and description of HLA-II motifs. Use of HLA-II gene-specific antibodies (i.e., pan-DR, pan-DP, or pan-DQ) may facilitate accurate motif deconvolution in such datasets.
Better Understanding of Antigen Presentation
While binding to HLA molecules is the most specific and best quantitatively characterized step of the antigen presentation process, it is likely that some additional filtering comes from cleavage in the proteasome, transport with TAP, and loading in the ER. As mentioned earlier, several recent studies suggest that including these additional parameters further improves prediction accuracy ( 70 , 71 , 159 , 166 ). One of the challenges there is to disentangle real biological signals from potential technical biases in MS data. Despite this caveat, it is likely that accumulating very large datasets of naturally presented HLA-I ligands is the only way to improve the accuracy of models of antigen presentation that go beyond the binding to HLA molecules. In addition, it could provide new information about how the HLA peptidome can be remodeled in response to extracellular signals, such as IFNγ stimulation ( 19 , 21 ). We, therefore, envision that screening how inhibition or activation of components of the antigen processing and presentation affect the nature of naturally presented HLA ligands on a large scale may reveal their role in shaping the HLA peptidome.
Non-Canonical HLA-I Ligands
Increasing evidences also suggest that non-canonical and cryptic peptides contribute to the HLA peptidome and expand the range of putative T cell epitopes. Laumont et al. have constructed a reference database of stop-to-stop translation products of six open reading frames of expressed RNAs and revealed that about 10% of the peptidome derive from presumably noncoding genomic sequences or exonic out-of-frame translation ( 87 ). Liepe et al. have reported that around 30% of the peptidome is derived from non-contiguous peptides spliced by the proteasome ( 169 ). Unexpectedly, spliced peptides displayed significantly lower predicted affinity than the normal peptides identified in the same samples ( 169 ) and did not show the expected HLA-I motifs. A very large database that is about two orders of magnitude larger than the typical protein-coding database was used to incorporate theoretical spliced products ( 169 ). Searching such large databases, especially in order to identify HLA peptides that have no enzymatic restrictions, may lead to improper control of false positives ( 170 ). In a recent preprint ( 171 ), we proposed an alternative, more conservative, approach to identify spliced peptides among HLA-I ligands based on de novo interpretation of high-quality spectra, suggesting that the number of such peptides may have been overestimated in the original study. The exact amount of spliced HLA-I ligands is still a matter of debate, and further studies will be needed to precisely estimate the fraction of spliced peptides actually displayed on HLA-I molecules. However, these potential issues suggest that putative spliced peptides may not all be appropriate for training HLA-I ligand predictors. Exploring non-canonical HLA ligands derived from translation of non-conventional regions in our genome or posttranslation events such as splicing is like finding a needle in a haystack. In silico predictions of such potential HLA ligands with existing tools may, therefore, lead to in-controlled numbers of false-positives, since the non-canonical space is theoretically orders of magnitude larger than the current canonical protein space. Hence, intensive proteogenomics based investigation of acquired HLA peptidomics data will likely play a central role in this endeavor and will require advanced computational tools and statistics to properly control for false positives.
Toward Predictions of Immunogenicity
Recent years have witnessed an unprecedented growth of in-depth and accurate MS data (Figure 1 ) that significantly enhanced our ability to predict antigen presentation. Unfortunately, these data cannot inform us about the most critical step in immune recognition, namely, the recognition of presented antigens by T cells. Much less is known there, and it is for instance, a disappointing fact that most predicted neoantigens from mutations found by exome sequencing of tumors are not recognized by T cells, although many resulting peptides do bind to HLA-I molecules. While direct identification of mutated peptides presented on the surface of cancer cells will likely improve the fraction of truly immunogenic epitopes ( 101 ), it is likely that many mutated peptides seen by MS will still not be immunogenic. Moreover, although binding affinity has been demonstrated to be useful for enriching pools of peptide in immunogenic epitopes (especially for class I), many known immunogenic epitopes display low-binding affinity, suggesting that they would be missed by approaches based on affinity predictions only. This is especially true for class II epitopes, where clear evidences indicate that different enzymes, peptide exchange mediates by HLA-DM or HLA-DO, pH gradients and peptide conformational flexibility play a role in selecting immunodominant epitopes ( 8 – 10 , 134 ). Unfortunately, currently, very little of this biological knowledge about class II antigen presentation could be used to improve predictions of class II epitopes.
Work by Calis et al. ( 172 ) suggested that some amino acids at non-anchor positions confer increased immunogenicity to HLA-I ligands. More recently, it has been observed that dissimilarity to self among mutated peptides predicted to have similar binding affinity as their wild-type counterpart can further help predicting immunogenic epitopes ( 173 ). Differences between the affinity of the wild-type and the mutated peptide, as well as stability of the MHC-I peptide interaction were also suggested to narrow down the list of immunogenic epitopes ( 174 ). Unfortunately, datasets of true immunogenic peptides from cancer or infectious diseases are still restricted to a few 100 peptides, limiting the power of machine learning approaches to infer properties of immunogenic epitopes ( 175 , 176 ). This is likely the main bottleneck toward our understanding of the determinants of immunogenicity. Therefore, recent high-throughput methods for screening T cells using for instance DNA barcoded multimers have the potential to provide critical information about the differences between immunogenic and non-immunogenic peptides ( 46 ). Importantly, most of these approaches require to select a priori the HLA ligands to be screened [with the exception of a recent phage display system ( 177 )]. Therefore, improved prediction of HLA ligands and antigen presentation will likely play an important role in optimizing the set of ligands currently tested for immunogenicity.
The first HLA-I motifs were described almost 30 years ago by looking at sequences obtained from MS analysis of eluted MHC-I ligands ( 89 , 90 ). Since then, much has been learned about HLA-I and HLA-II molecules through the analysis of their ligands. In human, this has resulted in a detailed description of HLA-I alleles binding specificities for the most common alleles and culminated with the development of pan-allele predictors. Recent years have witnessed an explosion of new high-quality data generated by MS about HLA-I ligands. Combined with advances in algorithms to analyze such data, this has led to refinement of known HLA-I motifs, discovery of new HLA-I motifs, characterization of peptide length distributions, analysis of N- and C-terminal extensions, characterization of antigen processing signals in flanking regions, analysis of the interplay between gene/protein expression, protein localization and peptide presentation, and evidences for presentation hotspots in the human proteome. For HLA-II ligands, MS studies have been recently used to study HLA-II motifs, suggesting that similar improvements may be observed there as well ( 21 , 138 – 140 ). Moreover, the current interest in neoantigen discovery will likely result in many more HLA peptidomics datasets from donors with diverse HLA backgrounds and different pathogeneses. This will provide unique opportunities to further improve our understanding of the rules of antigen presentation. To this end, it will be crucial that raw MS data are made publicly available, and that the reporting of HLA peptidomics data will comply with the recent minimal information about an Immuno-Peptidomics Experiment (MIAIPE) guidelines ( 178 ). Databases such as IEDB ( 25 ), PRIDE ( 179 ), or the SysteMHC Atlas ( 180 ) play a key role in this process, and it is our hope that soon all journals publishing HLA peptidomics studies will require deposition of the raw MS data in PRIDE and unfiltered lists of peptides in appropriate databases, or at least accessible in supplementary datasets.
Author Contributions
DG and MB-S designed the review and wrote the manuscript. DG analyzed the data and prepared the figures.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Julien Racle for insightful comments about the manuscript. DG is supported by the Swiss Cancer League (KFS-4104-02-2017-R).
Supplementary Material
The Supplementary Material for this article can be found online at https://www.frontiersin.org/articles/10.3389/fimmu.2018.01716/full#supplementary-material .
1. Neefjes J, Jongsma MLM, Paul P, Bakke O. Towards a systems understanding of MHC class I and MHC class II antigen presentation. Nat Rev Immunol (2011) 11:823–36. doi:10.1038/nri3084
PubMed Abstract | CrossRef Full Text | Google Scholar
2. Blum JS, Wearsch PA, Cresswell P. Pathways of antigen processing. Annu Rev Immunol (2013) 31:443–73. doi:10.1146/annurev-immunol-032712-095910
3. Vyas JM, Van der Veen AG, Ploegh HL. The known unknowns of antigen processing and presentation. Nat Rev Immunol (2008) 8:607–18. doi:10.1038/nri2368
4. Robinson J, Halliwell JA, Hayhurst JD, Flicek P, Parham P, Marsh SGE. The IPD and IMGT/HLA database: allele variant databases. Nucleic Acids Res (2015) 43:D423–31. doi:10.1093/nar/gku1161
5. Roche PA, Furuta K. The ins and outs of MHC class II-mediated antigen processing and presentation. Nat Rev Immunol (2015) 15:203–16. doi:10.1038/nri3818
6. Crotzer VL, Blum JS. Autophagy and its role in MHC-mediated antigen presentation. J Immunol (2009) 182:3335–41. doi:10.4049/jimmunol.0803458
7. Yin L, Maben ZJ, Becerra A, Stern LJ. Evaluating the role of HLA-DM in MHC class II-peptide association reactions. J Immunol (2015) 195:706–16. doi:10.4049/jimmunol.1403190
8. Kim A, Sadegh-Nasseri S. Determinants of immunodominance for CD4 T cells. Curr Opin Immunol (2015) 34:9–15. doi:10.1016/j.coi.2014.12.005
9. Sadegh-Nasseri S, Kim A. MHC class II auto-antigen presentation is unconventional. Front Immunol (2015) 6:372. doi:10.3389/fimmu.2015.00372
10. Sadegh-Nasseri S. A step-by-step overview of the dynamic process of epitope selection by major histocompatibility complex class II for presentation to helper T cells. F1000Res (2016) 5:1305. doi:10.12688/f1000research.7664.1
11. Johnson DB, Estrada MV, Salgado R, Sanchez V, Doxie DB, Opalenik SR, et al. Melanoma-specific MHC-II expression represents a tumour-autonomous phenotype and predicts response to anti-PD-1/PD-L1 therapy. Nat Commun (2016) 7:10582. doi:10.1038/ncomms10582
12. West MA, Wallin RPA, Matthews SP, Svensson HG, Zaru R, Ljunggren H-G, et al. Enhanced dendritic cell antigen capture via toll-like receptor-induced actin remodeling. Science (2004) 305:1153–7. doi:10.1126/science.1099153
13. Ackerman AL, Cresswell P. Cellular mechanisms governing cross-presentation of exogenous antigens. Nat Immunol (2004) 5:678–84. doi:10.1038/ni1082
14. Cruz FM, Colbert JD, Merino E, Kriegsman BA, Rock KL. The Biology and underlying mechanisms of cross-presentation of exogenous antigens on MHC-I molecules. Annu Rev Immunol (2017) 35:149–76. doi:10.1146/annurev-immunol-041015-055254
15. Bassani-Sternberg M, Bräunlein E, Klar R, Engleitner T, Sinitcyn P, Audehm S, et al. Direct identification of clinically relevant neoepitopes presented on native human melanoma tissue by mass spectrometry. Nat Commun (2016) 7:13404. doi:10.1038/ncomms13404
16. Caron E, Kowalewski DJ, Chiek Koh C, Sturm T, Schuster H, Aebersold R. Analysis of major histocompatibility complex (MHC) immunopeptidomes using mass spectrometry. Mol Cell Proteomics (2015) 14:3105–17. doi:10.1074/mcp.M115.052431
17. Vaughan K, Xu X, Caron E, Peters B, Sette A. Deciphering the MHC-associated peptidome: a review of naturally processed ligand data. Expert Rev Proteomics (2017) 14:729–36. doi:10.1080/14789450.2017.1361825
18. Bassani-Sternberg M, Pletscher-Frankild S, Jensen LJ, Mann M. Mass spectrometry of human leukocyte antigen class I peptidomes reveals strong effects of protein abundance and turnover on antigen presentation. Mol Cell Proteomics (2015) 14:658–73. doi:10.1074/mcp.M114.042812
19. Caron E, Vincent K, Fortier M-H, Laverdure J-P, Bramoullé A, Hardy M-P, et al. The MHC I immunopeptidome conveys to the cell surface an integrative view of cellular regulation. Mol Syst Biol (2011) 7:533. doi:10.1038/msb.2011.68
20. Fortier M-H, Caron E, Hardy M-P, Voisin G, Lemieux S, Perreault C, et al. The MHC class I peptide repertoire is molded by the transcriptome. J Exp Med (2008) 205:595–610. doi:10.1084/jem.20071985
21. Chong C, Marino F, Pak H, Racle J, Daniel RT, Müller M, et al. High-throughput and sensitive immunopeptidomics platform reveals profound interferonγ-mediated remodeling of the human leukocyte antigen (HLA) ligandome. Mol Cell Proteomics (2018) 17:533–48. doi:10.1074/mcp.TIR117.000383
22. Ferrington DA, Gregerson DS. Immunoproteasomes: structure, function, and antigen presentation. Prog Mol Biol Transl Sci (2012) 109:75–112. doi:10.1016/B978-0-12-397863-9.00003-1
23. Kincaid EZ, Murata S, Tanaka K, Rock KL. Specialized proteasome subunits have an essential role in the thymic selection of CD8(+) T cells. Nat Immunol (2016) 17:938–45. doi:10.1038/ni.3480
24. Robek MD, Garcia ML, Boyd BS, Chisari FV. Role of immunoproteasome catalytic subunits in the immune response to hepatitis B virus. J Virol (2007) 81:483–91. doi:10.1128/JVI.01779-06
25. Vita R, Overton JA, Greenbaum JA, Ponomarenko J, Clark JD, Cantrell JR, et al. The immune epitope database (IEDB) 3.0. Nucleic Acids Res (2015) 43:D405–12. doi:10.1093/nar/gku938
26. Weiskopf D, Yauch LE, Angelo MA, John DV, Greenbaum JA, Sidney J, et al. Insights into HLA-restricted T cell responses in a novel mouse model of dengue virus infection point toward new implications for vaccine design. J Immunol (2011) 187:4268–79. doi:10.4049/jimmunol.1101970
27. van der Bruggen P, Traversari C, Chomez P, Lurquin C, De Plaen E, Van den Eynde B, et al. A gene encoding an antigen recognized by cytolytic T lymphocytes on a human melanoma. Science (1991) 254:1643–7. doi:10.1126/science.1840703
28. Reuschenbach M, von Knebel DM, Wentzensen N. A systematic review of humoral immune responses against tumor antigens. Cancer Immunol Immunother (2009) 58:1535–44. doi:10.1007/s00262-009-0733-4
29. Simpson AJG, Caballero OL, Jungbluth A, Chen Y-T, Old LJ. Cancer/testis antigens, gametogenesis and cancer. Nat Rev Cancer (2005) 5:615–25. doi:10.1038/nrc1669
30. Bobisse S, Genolet R, Roberti A, Tanyi JL, Racle J, Stevenson BJ, et al. Sensitive and frequent identification of high avidity neo-epitope specific CD8 + T cells in immunotherapy-naive ovarian cancer. Nat Commun (2018) 9:1092. doi:10.1038/s41467-018-03301-0
CrossRef Full Text | Google Scholar
31. Carreno BM, Magrini V, Becker-Hapak M, Kaabinejadian S, Hundal J, Petti AA, et al. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science (2015) 348:803–8. doi:10.1126/science.aaa3828
32. Gros A, Parkhurst MR, Tran E, Pasetto A, Robbins PF, Ilyas S, et al. Prospective identification of neoantigen-specific lymphocytes in the peripheral blood of melanoma patients. Nat Med (2016) 22:433–8. doi:10.1038/nm.4051
33. Strønen E, Toebes M, Kelderman S, van Buuren MM, Yang W, van Rooij N, et al. Targeting of cancer neoantigens with donor-derived T cell receptor repertoires. Science (2016) 352:1337–41. doi:10.1126/science.aaf2288
34. Tran E, Turcotte S, Gros A, Robbins PF, Lu Y-C, Dudley ME, et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science (2014) 344:641–5. doi:10.1126/science.1251102
35. Yadav M, Jhunjhunwala S, Phung QT, Lupardus P, Tanguay J, Bumbaca S, et al. Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature (2014) 515:572–6. doi:10.1038/nature14001
36. Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med (2015) 372:2509–20. doi:10.1056/NEJMoa1500596
37. Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science (2015) 348:124–8. doi:10.1126/science.aaa1348
38. Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med (2014) 371:2189–99. doi:10.1056/NEJMoa1406498
39. Van Allen EM, Miao D, Schilling B, Shukla SA, Blank C, Zimmer L, et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science (2015) 350:207–11. doi:10.1126/science.aad0095
40. Tran E, Robbins PF, Lu Y-C, Prickett TD, Gartner JJ, Jia L, et al. T-cell transfer therapy targeting mutant KRAS in cancer. N Engl J Med (2016) 375:2255–62. doi:10.1056/NEJMoa1609279
41. Ott PA, Hu Z, Keskin DB, Shukla SA, Sun J, Bozym DJ, et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature (2017) 547:217–21. doi:10.1038/nature22991
42. Sahin U, Derhovanessian E, Miller M, Kloke B-P, Simon P, Löwer M, et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature (2017) 547:222–6. doi:10.1038/nature23003
43. Gfeller D, Bassani-Sternberg M, Schmidt J, Luescher IF. Current tools for predicting cancer-specific T cell immunity. Oncoimmunology (2016) 5:e1177691. doi:10.1080/2162402X.2016.1177691
44. Andersen RS, Thrue CA, Junker N, Lyngaa R, Donia M, Ellebæk E, et al. Dissection of T-cell antigen specificity in human melanoma. Cancer Res (2012) 72:1642–50. doi:10.1158/0008-5472.CAN-11-2614
45. Bentzen AK, Hadrup SR. Evolution of MHC-based technologies used for detection of antigen-responsive T cells. Cancer Immunol Immunother (2017) 66:657–66. doi:10.1007/s00262-017-1971-5
46. Bentzen AK, Marquard AM, Lyngaa R, Saini SK, Ramskov S, Donia M, et al. Large-scale detection of antigen-specific T cells using peptide-MHC-I multimers labeled with DNA barcodes. Nat Biotechnol (2016) 34:1037–45. doi:10.1038/nbt.3662
47. Linnemann C, van Buuren MM, Bies L, Verdegaal EME, Schotte R, Calis JJA, et al. High-throughput epitope discovery reveals frequent recognition of neo-antigens by CD4+ T cells in human melanoma. Nat Med (2015) 21:81–5. doi:10.1038/nm.3773
48. Elvin J, Cerundolo V, Elliott T, Townsend A. A quantitative assay of peptide-dependent class I assembly. Eur J Immunol (1991) 21:2025–31. doi:10.1002/eji.1830210909
49. Harndahl M, Rasmussen M, Røder G, Dalgaard Pedersen I, Sørensen M, Nielsen M, et al. Peptide-MHC class I stability is a better predictor than peptide affinity of CTL immunogenicity. Eur J Immunol (2012) 42:1405–16. doi:10.1002/eji.201141774
50. Sidney J, Assarsson E, Moore C, Ngo S, Pinilla C, Sette A, et al. Quantitative peptide binding motifs for 19 human and mouse MHC class I molecules derived using positional scanning combinatorial peptide libraries. Immunome Res (2008) 4:2. doi:10.1186/1745-7580-4-2
51. Townsend A, Elliott T, Cerundolo V, Foster L, Barber B, Tse A. Assembly of MHC class I molecules analyzed in vitro. Cell (1990) 62:285–95. doi:10.1016/0092-8674(90)90366-M
52. Wulf M, Hoehn P, Trinder P. Identification of human MHC class I binding peptides using the iTOPIA-epitope discovery system. Methods Mol Biol (2009) 524:361–7. doi:10.1007/978-1-59745-450-6_26
53. Bakker AH, Hoppes R, Linnemann C, Toebes M, Rodenko B, Berkers CR, et al. Conditional MHC class I ligands and peptide exchange technology for the human MHC gene products HLA-A1, -A3, -A11, and -B7. Proc Natl Acad Sci U S A (2008) 105:3825–30. doi:10.1073/pnas.0709717105
54. Hadrup SR, Toebes M, Rodenko B, Bakker AH, Egan DA, Ovaa H, et al. High-throughput T-cell epitope discovery through MHC peptide exchange. Methods Mol Biol (2009) 524:383–405. doi:10.1007/978-1-59745-450-6_28
55. Rodenko B, Toebes M, Hadrup SR, van Esch WJE, Molenaar AM, Schumacher TNM, et al. Generation of peptide-MHC class I complexes through UV-mediated ligand exchange. Nat Protoc (2006) 1:1120–32. doi:10.1038/nprot.2006.121
56. Sidney J, Southwood S, Moore C, Oseroff C, Pinilla C, Grey HM, et al. Measurement of MHC/peptide interactions by gel filtration or monoclonal antibody capture. Curr Protoc Immunol . (2013) 100:18.3.1–36. doi:10.1002/0471142735.im1803s100
57. Harndahl M, Rasmussen M, Røder G, Buus S. Real-time, high-throughput measurements of peptide-MHC-I dissociation using a scintillation proximity assay. J Immunol Methods (2011) 374:5–12. doi:10.1016/j.jim.2010.10.012
58. Rasmussen M, Harndahl M, Stryhn A, Boucherma R, Nielsen LL, Lemonnier FA, et al. Uncovering the peptide-binding specificities of HLA-C: a general strategy to determine the specificity of any MHC class I molecule. J Immunol (2014) 193:4790–802. doi:10.4049/jimmunol.1401689
59. Miles KM, Miles JJ, Madura F, Sewell AK, Cole DK. Real time detection of peptide-MHC dissociation reveals that improvement of primary MHC-binding residues can have a minimal, or no, effect on stability. Mol Immunol (2011) 48:728–32. doi:10.1016/j.molimm.2010.11.004
60. Justesen S, Harndahl M, Lamberth K, Nielsen L-LB, Buus S. Functional recombinant MHC class II molecules and high-throughput peptide-binding assays. Immunome Res (2009) 5:2. doi:10.1186/1745-7580-5-2
61. Salvat R, Moise L, Bailey-Kellogg C, Griswold KE. A high throughput MHC II binding assay for quantitative analysis of peptide epitopes. J Vis Exp (2014) (85):e51308. doi:10.3791/51308
62. Yin L, Stern LJ. Measurement of peptide binding to MHC class II molecules by fluorescence polarization. Curr Protoc Immunol . (2014) 106:5.10.1–12. doi:10.1002/0471142735.im0510s106
63. Gaseitsiwe S, Valentini D, Mahdavifar S, Reilly M, Ehrnst A, Maeurer M. Peptide microarray-based identification of Mycobacterium tuberculosis epitope binding to HLA-DRB1*0101, DRB1*1501, and DRB1*0401. Clin Vaccine Immunol (2010) 17:168–75. doi:10.1128/CVI.00208-09
64. Hammer J, Takacs B, Sinigaglia F. Identification of a motif for HLA-DR1 binding peptides using M13 display libraries. J Exp Med (1992) 176:1007–13. doi:10.1084/jem.176.4.1007
65. Jiang W, Boder ET. High-throughput engineering and analysis of peptide binding to class II MHC. Proc Natl Acad Sci U S A (2010) 107:13258–63. doi:10.1073/pnas.1006344107
66. Rajasagi M, Shukla SA, Fritsch EF, Keskin DB, DeLuca D, Carmona E, et al. Systematic identification of personal tumor-specific neoantigens in chronic lymphocytic leukemia. Blood (2014) 124:453–62. doi:10.1182/blood-2014-04-567933
67. Robbins PF, Lu Y-C, El-Gamil M, Li YF, Gross C, Gartner J, et al. Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nat Med (2013) 19:747–52. doi:10.1038/nm.3161
68. Castle JC, Kreiter S, Diekmann J, Löwer M, van de Roemer N, de Graaf J, et al. Exploiting the mutanome for tumor vaccination. Cancer Res (2012) 72:1081–91. doi:10.1158/0008-5472.CAN-11-3722
69. Kreiter S, Castle JC, Türeci Ö, Sahin U. Targeting the tumor mutanome for personalized vaccination therapy. Oncoimmunology (2012) 1:768–9. doi:10.4161/onci.19727
70. Abelin JG, Keskin DB, Sarkizova S, Hartigan CR, Zhang W, Sidney J, et al. Mass spectrometry profiling of HLA-associated peptidomes in mono-allelic cells enables more accurate epitope prediction. Immunity (2017) 46:315–26. doi:10.1016/j.immuni.2017.02.007
71. Pearson H, Daouda T, Granados DP, Durette C, Bonneil E, Courcelles M, et al. MHC class I-associated peptides derive from selective regions of the human genome. J Clin Invest (2016) 126:4690–701. doi:10.1172/JCI88590
72. Bassani-Sternberg M, Chong C, Guillaume P, Solleder M, Pak H, Gannon PO, et al. Deciphering HLA-I motifs across HLA peptidomes improves neo-antigen predictions and identifies allostery regulating HLA specificity. PLoS Comput Biol (2017) 13:e1005725. doi:10.1371/journal.pcbi.1005725
73. Mommen GPM, Frese CK, Meiring HD, van Gaans-van den Brink J, de Jong APJM, van Els CACM, et al. Expanding the detectable HLA peptide repertoire using electron-transfer/higher-energy collision dissociation (EThcD). Proc Natl Acad Sci U S A (2014) 111:4507–12. doi:10.1073/pnas.1321458111
74. Dargel C, Bassani-Sternberg M, Hasreiter J, Zani F, Bockmann J-H, Thiele F, et al. T cells engineered to express a T-cell receptor specific for glypican-3 to recognize and kill hepatoma cells in vitro and in mice. Gastroenterology (2015) 149:1042–52. doi:10.1053/j.gastro.2015.05.055
75. Di Marco M, Schuster H, Backert L, Ghosh M, Rammensee H-G, Stevanović S. Unveiling the peptide motifs of HLA-C and HLA-G from naturally presented peptides and generation of binding prediction matrices. J Immunol (2017) 199:2639–51. doi:10.4049/jimmunol.1700938
76. Gloger A, Ritz D, Fugmann T, Neri D. Mass spectrometric analysis of the HLA class I peptidome of melanoma cell lines as a promising tool for the identification of putative tumor-associated HLA epitopes. Cancer Immunol Immunother (2016) 65:1377–93. doi:10.1007/s00262-016-1897-3
77. Guasp P, Alvarez-Navarro C, Gomez-Molina P, Martín-Esteban A, Marcilla M, Barnea E, et al. The peptidome of Behçet’s disease-associated HLA-B*51:01 includes two subpeptidomes differentially shaped by endoplasmic reticulum aminopeptidase 1. Arthritis Rheumatol (2016) 68:505–15. doi:10.1002/art.39430
78. Hilton HG, McMurtrey CP, Han AS, Djaoud Z, Guethlein LA, Blokhuis JH, et al. The intergenic recombinant HLA-B*46:01 has a distinctive peptidome that includes KIR2DL3 ligands. Cell Rep (2017) 19:1394–405. doi:10.1016/j.celrep.2017.04.059
79. Jarmalavicius S, Welte Y, Walden P. High immunogenicity of the human leukocyte antigen peptidomes of melanoma tumor cells. J Biol Chem (2012) 287:33401–11. doi:10.1074/jbc.M112.358903
80. Kalaora S, Barnea E, Merhavi-Shoham E, Qutob N, Teer JK, Shimony N, et al. Use of HLA peptidomics and whole exome sequencing to identify human immunogenic neo-antigens. Oncotarget (2016) 7:5110–7. doi:10.18632/oncotarget.6960
81. Ritz D, Gloger A, Weide B, Garbe C, Neri D, Fugmann T. High-sensitivity HLA class I peptidome analysis enables a precise definition of peptide motifs and the identification of peptides from cell lines and patients’ sera. Proteomics (2016) 16:1570–80. doi:10.1002/pmic.201500445
82. Shraibman B, Kadosh DM, Barnea E, Admon A. Human leukocyte antigen (HLA) peptides derived from tumor antigens induced by inhibition of DNA methylation for development of drug-facilitated immunotherapy. Mol Cell Proteomics (2016) 15:3058–70. doi:10.1074/mcp.M116.060350
83. Singh-Jasuja H, Emmerich NPN, Rammensee H-G. The Tübingen approach: identification, selection, and validation of tumor-associated HLA peptides for cancer therapy. Cancer Immunol Immunother (2004) 53:187–95. doi:10.1007/s00262-003-0480-x
84. Weinschenk T, Gouttefangeas C, Schirle M, Obermayr F, Walter S, Schoor O, et al. Integrated functional genomics approach for the design of patient-individual antitumor vaccines. Cancer Res (2002) 62:5818–27.
PubMed Abstract | Google Scholar
85. Gubin MM, Zhang X, Schuster H, Caron E, Ward JP, Noguchi T, et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature (2014) 515:577–81. doi:10.1038/nature13988
86. Khodadoust MS, Olsson N, Wagar LE, Haabeth OAW, Chen B, Swaminathan K, et al. Antigen presentation profiling reveals recognition of lymphoma immunoglobulin neoantigens. Nature (2017) 543:723–7. doi:10.1038/nature21433
87. Laumont CM, Daouda T, Laverdure J-P, Bonneil E, Caron-Lizotte O, Hardy M-P, et al. Global proteogenomic analysis of human MHC class I-associated peptides derived from non-canonical reading frames. Nat Commun (2016) 7:10238. doi:10.1038/ncomms10238
88. Erhard F, Halenius A, Zimmermann C, L’Hernault A, Kowalewski DJ, Weekes MP, et al. Improved Ribo-seq enables identification of cryptic translation events. Nat Methods (2018) 4:e08890. doi:10.1038/nmeth.4631
89. Falk K, Rötzschke O, Stevanović S, Jung G, Rammensee H-G. Allele-specific motifs revealed by sequencing of self-peptides eluted from MHC molecules. Nature (1991) 351:290–6. doi:10.1038/351290a0
90. Hunt DF, Henderson RA, Shabanowitz J, Sakaguchi K, Michel H, Sevilir N, et al. Characterization of peptides bound to the class I MHC molecule HLA-A2.1 by mass spectrometry. Science (1992) 255:1261–3. doi:10.1126/science.1546328
91. Barnea E, Beer I, Patoka R, Ziv T, Kessler O, Tzehoval E, et al. Analysis of endogenous peptides bound by soluble MHC class I molecules: a novel approach for identifying tumor-specific antigens. Eur J Immunol (2002) 32:213–22. doi:10.1002/1521-4141(200201)32:1<213::AID-IMMU213>3.0.CO;2-8
92. Prilliman K, Lindsey M, Zuo Y, Jackson KW, Zhang Y, Hildebrand W. Large-scale production of class I bound peptides: assigning a signature to HLA-B*1501. Immunogenetics (1997) 45:379–85. doi:10.1007/s002510050219
93. Scull KE, Dudek NL, Corbett AJ, Ramarathinam SH, Gorasia DG, Williamson NA, et al. Secreted HLA recapitulates the immunopeptidome and allows in-depth coverage of HLA A*02:01 ligands. Mol Immunol (2012) 51:136–42. doi:10.1016/j.molimm.2012.02.117
94. Bassani-Sternberg M, Barnea E, Beer I, Avivi I, Katz T, Admon A. Soluble plasma HLA peptidome as a potential source for cancer biomarkers. Proc Natl Acad Sci U S A (2010) 107:18769–76. doi:10.1073/pnas.1008501107
95. Mobbs JI, Illing PT, Dudek NL, Brooks AG, Baker DG, Purcell AW, et al. The molecular basis for peptide repertoire selection in the human leucocyte antigen (HLA) C*06:02 molecule. J Biol Chem (2017) 292:17203–15. doi:10.1074/jbc.M117.806976
96. Schittenhelm RB, Dudek NL, Croft NP, Ramarathinam SH, Purcell AW. A comprehensive analysis of constitutive naturally processed and presented HLA-C*04:01 (Cw4)-specific peptides. Tissue Antigens (2014) 83:174–9. doi:10.1111/tan.12282
97. Trolle T, McMurtrey CP, Sidney J, Bardet W, Osborn SC, Kaever T, et al. The length distribution of class I-restricted T cell epitopes is determined by both peptide supply and MHC allele-specific binding preference. J Immunol (2016) 196:1480–7. doi:10.4049/jimmunol.1501721
98. Giam K, Ayala-Perez R, Illing PT, Schittenhelm RB, Croft NP, Purcell AW, et al. A comprehensive analysis of peptides presented by HLA-A1. Tissue Antigens (2015) 85:492–6. doi:10.1111/tan.12565
99. Yair-Sabag S, Tedeschi V, Vitulano C, Barnea E, Glaser F, Melamed Kadosh D, et al. The peptide repertoire of HLA-B27 may include ligands with lysine at P2 anchor position. Proteomics (2018) 18:e1700249. doi:10.1002/pmic.201700249
100. Schittenhelm RB, Sian TCCLK, Wilmann PG, Dudek NL, Purcell AW. Revisiting the arthritogenic peptide theory: quantitative not qualitative changes in the peptide repertoire of HLA-B27 allotypes. Arthritis Rheumatol (2015) 67:702–13. doi:10.1002/art.38963
101. Bassani-Sternberg M, Gfeller D. Unsupervised HLA peptidome deconvolution improves ligand prediction accuracy and predicts cooperative effects in peptide-HLA interactions. J Immunol (2016) 197:2492–9. doi:10.4049/jimmunol.1600808
102. Alvarez B, Barra C, Nielsen M, Andreatta M. Computational tools for the identification and interpretation of sequence motifs in immunopeptidomes. Proteomics (2018) 14:1700252. doi:10.1002/pmic.201700252
103. Andreatta M, Alvarez B, Nielsen M. GibbsCluster: unsupervised clustering and alignment of peptide sequences. Nucleic Acids Res (2017) 45:W458–63. doi:10.1093/nar/gkx248
104. Kim Y, Sidney J, Pinilla C, Sette A, Peters B. Derivation of an amino acid similarity matrix for peptide: MHC binding and its application as a Bayesian prior. BMC Bioinformatics (2009) 10:394. doi:10.1186/1471-2105-10-394
105. Lund O, Nielsen M, Lundegaard C, Keşmir C, Brunak S. Immunological Bioinformatics . MIT Press (2005).
Google Scholar
106. Rammensee H, Bachmann J, Emmerich NP, Bachor OA, Stevanović S. SYFPEITHI: database for MHC ligands and peptide motifs. Immunogenetics (1999) 50:213–9. doi:10.1007/s002510050595
107. Zhang H, Lund O, Nielsen M. The PickPocket method for predicting binding specificities for receptors based on receptor pocket similarities: application to MHC-peptide binding. Bioinformatics (2009) 25:1293–9. doi:10.1093/bioinformatics/btp137
108. Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res (1997) 25:3389–402. doi:10.1093/nar/25.17.3389
109. Henikoff S, Henikoff JG. Amino acid substitution matrices from protein blocks. Proc Natl Acad Sci U S A (1992) 89:10915–9. doi:10.1073/pnas.89.22.10915
110. Andreatta M, Nielsen M. Gapped sequence alignment using artificial neural networks: application to the MHC class I system. Bioinformatics (2016) 32:511–7. doi:10.1093/bioinformatics/btv639
111. Jørgensen KW, Rasmussen M, Buus S, Nielsen M. NetMHCstab – predicting stability of peptide-MHC-I complexes; impacts for cytotoxic T lymphocyte epitope discovery. Immunology (2014) 141:18–26. doi:10.1111/imm.12160
112. Nielsen M, Lundegaard C, Worning P, Lauemøller SL, Lamberth K, Buus S, et al. Reliable prediction of T-cell epitopes using neural networks with novel sequence representations. Protein Sci (2003) 12:1007–17. doi:10.1110/ps.0239403
113. Rubinsteyn A, O’Donnell T, Damaraju N, Hammerbacher J. Predicting peptide-MHC binding affinities with imputed training data. bioRxiv (2016) 054775. doi:10.1101/054775
114. Vang YS, Xie X. HLA class I binding prediction via convolutional neural networks. Bioinformatics (2017) 33:2658–65. doi:10.1093/bioinformatics/btx264
115. Peters B, Tong W, Sidney J, Sette A, Weng Z. Examining the independent binding assumption for binding of peptide epitopes to MHC-I molecules. Bioinformatics (2003) 19:1765–72. doi:10.1093/bioinformatics/btg247
116. Han Y, Kim D. Deep convolutional neural networks for pan-specific peptide-MHC class I binding prediction. BMC Bioinformatics (2017) 18:585. doi:10.1186/s12859-017-1997-x
117. Jurtz V, Paul S, Andreatta M, Marcatili P, Peters B, Nielsen M. NetMHCpan-4.0: improved peptide-MHC class I interaction predictions integrating eluted ligand and peptide binding affinity data. J Immunol (2017) 199:3360–8. doi:10.4049/jimmunol.1700893
118. Rasmussen M, Fenoy E, Harndahl M, Kristensen AB, Nielsen IK, Nielsen M, et al. Pan-specific prediction of peptide-MHC class I complex stability, a correlate of T cell immunogenicity. J Immunol (2016) 197:1517–24. doi:10.4049/jimmunol.1600582
119. Nielsen M, Connelley T, Ternette N. Improved prediction of Bovine Leucocyte Antigens (BoLA) presented ligands by use of mass spectrometry-determined ligand- and in-vitro binding data. J Proteome Res (2017) 17(1):559–67. doi:10.1021/acs.jproteome.7b00675
120. Guillaume P, Picaud S, Baumgaertner P, Montandon N, Schmidt J, Speiser DE, et al. The C-terminal extension landscape of naturally presented HLA-I ligands. Proc Natl Acad Sci U S A (2018) 115:5083–8. doi:10.1073/pnas.1717277115
121. Gfeller D. Uncovering new aspects of protein interactions through analysis of specificity landscapes in peptide recognition domains. FEBS Lett (2012) 586:2764–72. doi:10.1016/j.febslet.2012.03.054
122. Gfeller D, Butty F, Wierzbicka M, Verschueren E, Vanhee P, Huang H, et al. The multiple-specificity landscape of modular peptide recognition domains. Mol Syst Biol (2011) 7:484. doi:10.1038/msb.2011.18
123. Kim T, Tyndel MS, Huang H, Sidhu SS, Bader GD, Gfeller D, et al. MUSI: an integrated system for identifying multiple specificity from very large peptide or nucleic acid data sets. Nucleic Acids Res (2012) 40:e47. doi:10.1093/nar/gkr1294
124. Andreatta M, Lund O, Nielsen M. Simultaneous alignment and clustering of peptide data using a Gibbs sampling approach. Bioinformatics (2013) 29:8–14. doi:10.1093/bioinformatics/bts621
125. Storkus WJ, Zeh HJ, Salter RD, Lotze MT. Identification of T-cell epitopes: rapid isolation of class I-presented peptides from viable cells by mild acid elution. J Immunother Emphasis Tumor Immunol (1993) 14:94–103. doi:10.1097/00002371-199308000-00003
126. Sugawara S, Abo T, Kumagai K. A simple method to eliminate the antigenicity of surface class I MHC molecules from the membrane of viable cells by acid treatment at pH 3. J Immunol Methods (1987) 100:83–90. doi:10.1016/0022-1759(87)90175-X
127. Lanoix J, Durette C, Courcelles M, Cossette É, Comtois-Marotte S, Hardy M-P, et al. Comparison of the MHC I immunopeptidome repertoire of B-cell lymphoblasts using two isolation methods. Proteomics (2018) 18(12):e1700251. doi:10.1002/pmic.201700251
128. Nielsen M, Lund O. NN-align. An artificial neural network-based alignment algorithm for MHC class II peptide binding prediction. BMC Bioinformatics (2009) 10:296. doi:10.1186/1471-2105-10-296
129. Jensen KK, Andreatta M, Marcatili P, Buus S, Greenbaum JA, Yan Z, et al. Improved methods for predicting peptide binding affinity to MHC class II molecules. Immunology (2018) 154:394–406. doi:10.1111/imm.12889
130. Singh H, Raghava GP. ProPred: prediction of HLA-DR binding sites. Bioinformatics (2001) 17:1236–7. doi:10.1093/bioinformatics/17.12.1236
131. Guan P, Doytchinova IA, Zygouri C, Flower DR. MHCPred: a server for quantitative prediction of peptide-MHC binding. Nucleic Acids Res (2003) 31:3621–4. doi:10.1093/nar/gkg510
132. Sturniolo T, Bono E, Ding J, Raddrizzani L, Tuereci O, Sahin U, et al. Generation of tissue-specific and promiscuous HLA ligand databases using DNA microarrays and virtual HLA class II matrices. Nat Biotechnol (1999) 17:555–61. doi:10.1038/9858
133. Wang P, Sidney J, Dow C, Mothé B, Sette A, Peters B. A systematic assessment of MHC class II peptide binding predictions and evaluation of a consensus approach. PLoS Comput Biol (2008) 4:e1000048. doi:10.1371/journal.pcbi.1000048
134. Yin L, Stern LJ. HLA-DM focuses on conformational flexibility around P1 pocket to catalyze peptide exchange. Front Immunol (2013) 4:336. doi:10.3389/fimmu.2013.00336
135. Vogt AB, Kropshofer H, Kalbacher H, Kalbus M, Rammensee H-G, Coligan JE, et al. Ligand motifs of HLA-DRB5*0101 and DRB1*1501 molecules delineated from self-peptides. J Immunol (1994) 153:1665–73.
136. Sofron A, Ritz D, Neri D, Fugmann T. High-resolution analysis of the murine MHC class II immunopeptidome. Eur J Immunol (2015) 46:319–28. doi:10.1002/eji.201545930
137. Fugmann T, Sofron A, Ritz D, Bootz F, Neri D. The MHC class II immunopeptidome of lymph nodes in health and in chemically induced colitis. Journal Immunol (2016) 198:1357–64. doi:10.4049/jimmunol.1601157
138. Ooi JD, Petersen J, Tan YH, Huynh M, Willett ZJ, Ramarathinam SH, et al. Dominant protection from HLA-linked autoimmunity by antigen-specific regulatory T cells. Nature (2017) 545:243–7. doi:10.1038/nature22329
139. Ritz D, Sani E, Debiec H, Ronco P, Neri D, Fugmann T. Membranal and blood-soluble HLA class II peptidome analyses using data-dependent and independent acquisition. Proteomics (2018) 34:1700246. doi:10.1002/pmic.201700246
140. Barra CM, Alvarez B, Andreatta M, Buus S, Nielsen M. Footprints of antigen processing boost MHC class II natural ligand binding predictions. bioRxiv (2018):285767. doi:10.1101/285767
141. Mommen GPM, Marino F, Meiring HD, Poelen MCM, van Gaans-van den Brink JAM, Mohammed S, et al. Sampling from the proteome to the human leukocyte antigen-DR (HLA-DR) ligandome proceeds via high specificity. Mol Cell Proteomics (2016) 15:1412–23. doi:10.1074/mcp.M115.055780
142. Hassan C, Chabrol E, Jahn L, Kester MGD, de Ru AH, Drijfhout JW, et al. Naturally processed non-canonical HLA-A*02:01 presented peptides. J Biol Chem (2015) 290:2593–603. doi:10.1074/jbc.M114.607028
143. Gfeller D, Guillaume P, Michaux J, Pak H-S, Daniel RT, Racle J, et al. Peptide length distribution and multiple specificity in naturally presented HLA-I ligands. bioRxiv (2018):335661. doi:10.1101/335661
144. Collins EJ, Garboczi DN, Wiley DC. Three-dimensional structure of a peptide extending from one end of a class I MHC binding site. Nature (1994) 371:626–9. doi:10.1038/371626a0
145. Tenzer S, Wee E, Burgevin A, Stewart-Jones G, Friis L, Lamberth K, et al. Antigen processing influences HIV-specific cytotoxic T lymphocyte immunodominance. Nat Immunol (2009) 10:636–46. doi:10.1038/ni.1728
146. McMurtrey C, Trolle T, Sansom T, Remesh SG, Kaever T, Bardet W, et al. Toxoplasma gondii peptide ligands open the gate of the HLA class I binding groove. Elife (2016) 5:246. doi:10.7554/eLife.12556
147. Remesh SG, Andreatta M, Ying G, Kaever T, Nielsen M, McMurtrey C, et al. Unconventional peptide presentation by major histocompatibility complex (MHC) class I Allele HLA-A*02:01: BREAKING CONFINEMENT. J Biol Chem (2017) 292:5262–70. doi:10.1074/jbc.M117.776542
148. Pymm P, Illing PT, Ramarathinam SH, O’Connor GM, Hughes VA, Hitchen C, et al. MHC-I peptides get out of the groove and enable a novel mechanism of HIV-1 escape. Nat Struct Mol Biol (2017) 219:277. doi:10.1038/nsmb.3381
149. Li X, Lamothe PA, Walker BD, Wang J-H. Crystal structure of HLA-B*5801 with a TW10 HIV Gag epitope reveals a novel mode of peptide presentation. Cell Mol Immunol (2017) 14:631–4. doi:10.1038/cmi.2017.24
150. Alpízar A, Marino F, Ramos-Fernández A, Lombardía M, Jeko A, Pazos F, et al. A molecular basis for the presentation of phosphorylated peptides by HLA-B antigens. Mol Cell Proteomics (2016) 16(2):181–93. doi:10.1074/mcp.M116.063800
151. Marino F, Bern M, Mommen GPM, Leney AC, van Gaans-van den Brink JAM, Bonvin AMJJ, et al. Extended O-GlcNAc on HLA class-I-bound peptides. J Am Chem Soc (2015) 137:10922–5. doi:10.1021/jacs.5b06586
152. Marino F, Mommen GPM, Jeko A, Meiring HD, van Gaans-van den Brink JAM, Scheltema RA, et al. Arginine (Di)methylated human leukocyte antigen class I peptides are favorably presented by HLA-B*07. J Proteome Res (2017) 16:34–44. doi:10.1021/acs.jproteome.6b00528
153. Andersen MH, Bonfill JE, Neisig A, Arsequell G, Sondergaard I, Valencia G, et al. Phosphorylated peptides can be transported by TAP molecules, presented by class I MHC molecules, and recognized by phosphopeptide-specific CTL. J Immunol (1999) 163:3812–8.
154. Petersen J, Wurzbacher SJ, Williamson NA, Ramarathinam SH, Reid HH, Nair AKN, et al. Phosphorylated self-peptides alter human leukocyte antigen class I-restricted antigen presentation and generate tumor-specific epitopes. Proc Natl Acad Sci U S A (2009) 106:2776–81. doi:10.1073/pnas.0812901106
155. Zarling AL, Polefrone JM, Evans AM, Mikesh LM, Shabanowitz J, Lewis ST, et al. Identification of class I MHC-associated phosphopeptides as targets for cancer immunotherapy. Proc Natl Acad Sci U S A (2006) 103:14889–94. doi:10.1073/pnas.0604045103
156. Ciudad MT, Sorvillo N, van Alphen FP, Catalán D, Meijer AB, Voorberg J, et al. Analysis of the HLA-DR peptidome from human dendritic cells reveals high affinity repertoires and nonconventional pathways of peptide generation. J Leukoc Biol (2017) 101:15–27. doi:10.1189/jlb.6HI0216-069R
157. Collado JA, Alvarez I, Ciudad MT, Espinosa G, Canals F, Pujol-Borrell R, et al. Composition of the HLA-DR-associated human thymus peptidome. Eur J Immunol (2013) 43:2273–82. doi:10.1002/eji.201243280
158. Clement CC, Becerra A, Yin L, Zolla V, Huang L, Merlin S, et al. The dendritic cell major histocompatibility complex II (MHC II) peptidome derives from a variety of processing pathways and includes peptides with a broad spectrum of HLA-DM sensitivity. J Biol Chem (2016) 291:5576–95. doi:10.1074/jbc.M115.655738
159. Müller M, Gfeller D, Coukos G, Bassani-Sternberg M. “Hotspots” of antigen presentation revealed by human leukocyte antigen ligandomics for neoantigen prioritization. Front Immunol (2017) 8:1367. doi:10.3389/fimmu.2017.01367
160. Dengjel J, Schoor O, Fischer R, Reich M, Kraus M, Müller M, et al. Autophagy promotes MHC class II presentation of peptides from intracellular source proteins. Proc Natl Acad Sci U S A (2005) 102:7922–7. doi:10.1073/pnas.0501190102
161. Kropshofer H, Max H, Halder T, Kalbus M, Muller CA, Kalbacher H. Self-peptides from four HLA-DR alleles share hydrophobic anchor residues near the NH2-terminal including proline as a stop signal for trimming. J Immunol (1993) 151:4732–42.
162. Godkin AJ, Smith KJ, Willis A, Tejada-Simon MV, Zhang J, Elliott T, et al. Naturally processed HLA class II peptides reveal highly conserved immunogenic flanking region sequence preferences that reflect antigen processing rather than peptide-MHC interactions. J Immunol (2001) 166:6720–7. doi:10.4049/jimmunol.166.11.6720
163. Andreatta M, Jurtz VI, Kaever T, Sette A, Peters B, Nielsen M. Machine learning reveals a non-canonical mode of peptide binding to MHC class II molecules. Immunology (2017) 152:255–64. doi:10.1111/imm.12763
164. Nielsen M, Lundegaard C, Lund O, Keşmir C. The role of the proteasome in generating cytotoxic T-cell epitopes: insights obtained from improved predictions of proteasomal cleavage. Immunogenetics (2005) 57:33–41. doi:10.1007/s00251-005-0781-7
165. Doytchinova IA, Guan P, Flower DR. EpiJen: a server for multistep T cell epitope prediction. BMC Bioinformatics (2006) 7:131. doi:10.1186/1471-2105-7-131
166. Tenzer S, Peters B, Bulik S, Schoor O, Lemmel C, Schatz MM, et al. Modeling the MHC class I pathway by combining predictions of proteasomal cleavage, TAP transport and MHC class I binding. Cell Mol Life Sci (2005) 62:1025–37. doi:10.1007/s00018-005-4528-2
167. Zhang GL, Petrovsky N, Kwoh CK, August JT, Brusic V. PRED(TAP): a system for prediction of peptide binding to the human transporter associated with antigen processing. Immunome Res (2006) 2:3. doi:10.1186/1745-7580-2-3
168. Jappe EC, Kringelum J, Trolle T, Nielsen M. Predicted MHC peptide binding promiscuity explains MHC class I “hotspots” of antigen presentation defined by mass spectrometry eluted ligand data. Immunology (2018) 154:407–17. doi:10.1111/imm.12905
169. Liepe J, Marino F, Sidney J, Jeko A, Bunting DE, Sette A, et al. A large fraction of HLA class I ligands are proteasome-generated spliced peptides. Science (2016) 354:354–8. doi:10.1126/science.aaf4384
170. Nesvizhskii AI. Proteogenomics: concepts, applications and computational strategies. Nat Methods (2014) 11:1114–25. doi:10.1038/nmeth.3144
171. Mylonas R, Beer I, Iseli C, Chong C, Pak H, Gfeller D, et al. Estimating the contribution of proteasomal spliced peptides to the HLA-I ligandome. bioRxiv (2018):288209. doi:10.1101/288209
172. Calis JJA, Maybeno M, Greenbaum JA, Weiskopf D, De Silva AD, Sette A, et al. Properties of MHC class I presented peptides that enhance immunogenicity. PLoS Comput Biol (2013) 9:e1003266. doi:10.1371/journal.pcbi.1003266
173. Bjerregaard A-M, Nielsen M, Jurtz V, Barra CM, Hadrup SR, Szallasi Z, et al. An analysis of natural T cell responses to predicted tumor neoepitopes. Front Immunol (2017) 8:1566. doi:10.3389/fimmu.2017.01566
174. Duan F, Duitama J, Al Seesi S, Ayres CM, Corcelli SA, Pawashe AP, et al. Genomic and bioinformatic profiling of mutational neoepitopes reveals new rules to predict anticancer immunogenicity. J Exp Med (2014) 211:2231–48. doi:10.1084/jem.20141308
175. van Buuren MM, Calis JJ, Schumacher TN. High sensitivity of cancer exome-based CD8 T cell neo-antigen identification. Oncoimmunology (2014) 3:e28836. doi:10.4161/onci.28836
176. Fritsch EF, Rajasagi M, Ott PA, Brusic V, Hacohen N, Wu CJ. HLA-binding properties of tumor neoepitopes in humans. Cancer Immunol Res (2014) 2:522–9. doi:10.1158/2326-6066.CIR-13-0227
177. Gee MH, Han A, Lofgren SM, Beausang JF, Mendoza JL, Birnbaum ME, et al. Antigen identification for orphan t cell receptors expressed on tumor-infiltrating lymphocytes. Cell (2018) 172:549.e–63.e. doi:10.1016/j.cell.2017.11.043
178. Lill JR, van Veelen PA, Tenzer S, Admon A, Caron E, Elias J, et al. Minimal information about an immuno-peptidomics experiment (MIAIPE). Proteomics (2018) 18(12):e1800110. doi:10.1002/pmic.201800110
179. Vizcaíno JA, Csordas A, Del-Toro N, Dianes JA, Griss J, Lavidas I, et al. 2016 update of the PRIDE database and its related tools. Nucleic Acids Res (2016) 44:11033–11033. doi:10.1093/nar/gkv1145
180. Shao W, Pedrioli PGA, Wolski W, Scurtescu C, Schmid E, Vizcaíno JA, et al. The SysteMHC Atlas project. Nucleic Acids Res (2018) 46:D1237–47. doi:10.1093/nar/gkx664
181. Karosiene E, Lundegaard C, Lund O, Nielsen M. NetMHCcons: a consensus method for the major histocompatibility complex class I predictions. Immunogenetics (2012) 64:177–86. doi:10.1007/s00251-011-0579-8
182. Moutaftsi M, Peters B, Pasquetto V, Tscharke DC, Sidney J, Bui H-H, et al. A consensus epitope prediction approach identifies the breadth of murine T(CD8+)-cell responses to vaccinia virus. Nat Biotechnol (2006) 24:817–9. doi:10.1038/nbt1215
183. Bhattacharya R, Sivakumar A, Tokheim C, Guthrie VB, Anagnostou V, Velculescu VE, et al. Evaluation of machine learning methods to predict peptide binding to MHC Class I proteins. bioRxiv (2017) 154757. doi:10.1101/154757
184. Liu G, Li D, Li Z, Qiu S, Li W, Chao C-C, et al. PSSMHCpan: a novel PSSM-based software for predicting class I peptide-HLA binding affinity. GigaScience (2017) 6:1–11. doi:10.1093/gigascience/gix017
185. Sidney J, Peters B, Frahm N, Brander C, Sette A. HLA class I supertypes: a revised and updated classification. BMC Immunol (2008) 9:1. doi:10.1186/1471-2172-9-1
Keywords: human leukocyte antigen peptidomics, human leukocyte antigen ligand prediction, antigen presentation, T cell epitope, computational immunology
Citation: Gfeller D and Bassani-Sternberg M (2018) Predicting Antigen Presentation—What Could We Learn From a Million Peptides? Front. Immunol. 9:1716. doi: 10.3389/fimmu.2018.01716
Received: 27 April 2018; Accepted: 12 July 2018; Published: 25 July 2018
Reviewed by:
Copyright: © 2018 Gfeller and Bassani-Sternberg. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY) . The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: David Gfeller, david.gfeller@unil.ch ; Michal Bassani-Sternberg, michal.bassani@chuv.ch
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
If you're seeing this message, it means we're having trouble loading external resources on our website.
If you're behind a web filter, please make sure that the domains *.kastatic.org and *.kasandbox.org are unblocked.
To log in and use all the features of Khan Academy, please enable JavaScript in your browser.
Biology library
Course: biology library > unit 33.
- Role of phagocytes in innate or nonspecific immunity
- Types of immune responses: Innate and adaptive, humoral vs. cell-mediated
- B lymphocytes (B cells)
Professional antigen presenting cells (APC) and MHC II complexes
- Helper T cells
- Cytotoxic T cells and MHC I complexes
- Review of B cells, CD4+ T cells and CD8+ T cells
- Inflammatory response
Want to join the conversation?
- Upvote Button navigates to signup page
- Downvote Button navigates to signup page
- Flag Button navigates to signup page

Video transcript

You have no alerts at this time
- Product Catalog
- Member Directory

Patient Care and Research
- Guidelines and Consensus Documents
- Patient Safety
- Funding Opportunities
- Scientific Challenges
- Professional Development
- Provider Resources

An official website of the United States government
The .gov means it’s official. Federal government websites often end in .gov or .mil. Before sharing sensitive information, make sure you’re on a federal government site.
The site is secure. The https:// ensures that you are connecting to the official website and that any information you provide is encrypted and transmitted securely.
- Publications
- Account settings
Preview improvements coming to the PMC website in October 2024. Learn More or Try it out now .
- Advanced Search
- Journal List
- Front Immunol
Antigen Presentation in the Lung
The lungs are constantly exposed to environmental and infectious agents such as dust, viruses, fungi, and bacteria that invade the lungs upon breathing. The lungs are equipped with an immune defense mechanism that involves a wide variety of immunological cells to eliminate these agents. Various types of dendritic cells (DCs) and macrophages (MACs) function as professional antigen-presenting cells (APCs) that engulf pathogens through endocytosis or phagocytosis and degrade proteins derived from them into peptide fragments. During this process, DCs and MACs present the peptides on their major histocompatibility complex class I (MHC-I) or MHC-II protein complex to naïve CD8 + or CD4 + T cells, respectively. In addition to these cells, recent evidence supports that antigen-specific effector and memory T cells are activated by other lung cells such as endothelial cells, epithelial cells, and monocytes through antigen presentation. In this review, we summarize the molecular mechanisms of antigen presentation by APCs in the lungs and their contribution to immune response.
Introduction
The lung is the peripheral tissue that exchanges gas during respiration; therefore, it is exposed to the outer environment, which potentially increases the risk of invasion by viral and bacterial pathogens. Respiratory viruses, including influenza virus and recent coronavirus, induce inflammation and tissue damage, leading to disorders of the lungs. The high infectivity and spreadability of these viruses have caused a worldwide pandemic in recent years and has provoked the argument for recurrent infection and efficacy of vaccination in order to suppress the pandemic. Innate immune cells such as dendritic cells (DCs) and macrophages (MACs) in the lungs form the first line of defense by recognizing the molecular structures common to pathogens, called pathogen-associated molecular patterns, through pattern recognition receptors ( 1 , 2 ). During the past decade, various types of lung DCs and MACs have been identified and classified according to surface markers, expression genes, and corresponding transcription factors with specialized functions. These DCs and MACs function as antigen-presenting cells (APCs) that engulf pathogens through endocytosis or phagocytosis and present their peptides on major histocompatibility complex class I (MHC-I) or MHC-II protein complex to naïve CD8 + or CD4 + T cells, respectively. Although DCs and MACs are known as professional APCs with a higher expression of co-stimulatory molecules, such as CD80 and CD86, other types of cells such as monocytes and epithelial cells in the lungs also have the potential to present antigens to T cells.
APCs load peptides derived from exogenous antigens on MHC-II and present peptide-MHC-II complex to CD4 + T cells whereas APCs load peptides derived from both endogenous and cytosolic antigens on MHC-I and present peptide-MHC-I complex to CD8 + T cells ( Figures 1 , 2 ). In addition, specific APCs take up exogenous antigens, process them, and load peptides onto MHC-I to CD8 + T cells, a process called antigen cross-presentation ( 3 ). Lung DCs are largely divided into three major subsets: cDC1s, cDC2s, and plasmacytoid DCs (pDCs). These DCs have been focused on as key regulators of T cell responses ( 4 ); however, recent evidence indicates that other types of cells in the lung, such as MACs, monocytes, and epithelial cells, also have antigen presentation capacity to both CD4 + and CD8 + T cells. MACs in the lung are mainly classified into alveolar macrophages (AMs) and interstitial macrophages (IMs). Lung epithelial cells (LECs) consist of alveolar type I (ATI) and alveolar type II (ATII) cells in the alveoli, and the predominant cell types constituting the bronchial airway epithelium include endothelial cells, basal progenitor cells, ciliated cells, secretory club cells, and goblet cells ( 5 , 6 ). Lung DCs, MAC and LECs express MHC-I and/or MHC-II on their cell surface and potentially present antigen to CD4 + or CD8 + T cells ( 7 ).

Antigen presentation on MHC-II molecule. Extracellular antigens are endocytosed or phagocytosed, and intracellular antigens are translocated to the late-endosome or the lysosome via autophagosome- or LAMP-2A- mediated autophagy. Then these antigens are degraded by asparaginyl endopeptidase and cathepsin. MHC-II is synthesized in ER and mainly pooled at the plasma membrane as MHC-II-Ii chain complex. When the complex translocates from the ER or the plasma membrane to the acidic compartment, Ii chain is degraded into CLIP and driven out by interaction with H2-M. Afterward, antigen peptides bind to the MHC-II and the peptide-MHC-II complex exports to the cell surface.

Antigen cross-presentation on MHC-I molecule. Extracellular antigens are presented via “vacuolar pathway” or “cytosolic pathway” in the cross-presentation pathway. In the vacuolar pathway, endocytosed antigen peptides are degraded by cathepsin S and bind to MHC-I in the endosomal compartment. In the cytosolic pathway, endocytosed or phagocytosed extracellular antigens are translocated to the cytosol via Sec61 and degraded by proteasome. The degraded peptides are transported into the ER or the endosome via TAP and trimmed by ERAP (in the ER) or IRAP (in the endosomes). TAP form PLC with MHC-I, ERp57 and calreticulin. Afterward, the trimmed peptides bind to the MHC-I and transported to the cell surface. The MHC-I in the endosomes is recruited from the plasma membrane through Rab11a + recycle endosome, the ER, or the ERGIC. Antigen degradation regulated by the acidification in the endosome, the phagosome, and the lysosome by V-ATPase. On the other hand, NADPH oxidase NOX2 regulates phagosomal alkalization and is recruited to the phagosomes by Rab27a-dependent pathway.
During pathogen infection in the lung, pathogen-specific CD4 + and CD8 + cells are primed in the lung-draining lymph nodes by antigen-presenting DCs that migrate from the infected area in the lung ( 8 , 9 ). Antigen-presenting DCs encounter naïve CD4 + and CD8 + T cells in the lymph nodes, where antigen-specific T cells are selected, and the proliferation and differentiation by antigen presentation on MHC molecules are induced along with the assistance of co-stimulatory molecules and the local cytokine environment ( 10 , 11 ). Antigen-specific CD4 + and CD8 + T cells in the lymph nodes migrate to the lungs to directly eliminate infected cells or induce the accumulation of other immunological cells for pathogen clearance. In addition, antigen-specific T cells encounter local APCs in the lungs, including DCs, MACs, monocytes, and LECs, and further differentiate and expand in the lung ( 12 ). Parts of antigen-specific cells differentiate into long-lived memory cells, which are divided into three types of population: central memory T (T CM ) cells, which are largely found in secondary lymphoid organs; effector memory T (T EF ) cells, which systematically circulate, transiently entering peripheral tissue, and resident memory T (T RM ) cells, a non-circulating, self-renewing population located in peripheral tissues including the lungs ( 13 , 14 ). There has been increasing evidence that antigen-specific memory T cell formation through antigen presentation or cytokines is facilitated by various types of lung cells. In this review, we summarize the molecular mechanisms of antigen presentation to MHC-I and MHC-II on APCs and memory T cell formation by APCs during pathogen infection in the lung.
Molecular Basis of Antigen Presentation to CD4 + T Cells
In general, extracellular antigens are endocytosed or phagocytosed by APCs and degraded by proteases such as asparaginyl endopeptidase ( 15 ) and cathepsins S, B, H, and L ( 16 – 18 ). Degraded peptides are ultimately presented on MHC-II molecules to prime CD4 + T cells ( 19 ) ( Figure 1 ). However, less than 30% of antigens on MHC-II are derived from endogenous antigens, such as cytoplasmic or nuclear antigens ( 20 , 21 ). Regardless of peptides derived from self or non-self-antigens, these peptides can be presented by APCs, non-professional APCs, or tumor cells mainly via autophagosome- or chaperone-mediated autophagy ( 22 ). Antigen degradation is mediated by the fusion of autophagosomes with endosomes and lysosomes in autophagosome-mediated autophagy ( 23 ). Antigens degraded by the proteasome in the cytosol are translocated to the late endosome or lysosome, which is enhanced by lysosome-associated membrane protein 2A (LAMP-2A) ( 24 ).
Newly synthesized MHC-II forms a complex with the invariant (Ii) chain in the endoplasmic reticulum (ER), and is pooled in the ER or plasma membrane and then respectively, translocated to the endosomes and lysosomes either directly ( 25 ) or indirectly though endocytosis ( 26 , 27 ); however, the complex cannot bind to antigen peptides ( 28 , 29 ). The Ii chain is degraded into a small fragment called class II-associated Ii chain peptide (CLIP) and binds to MHC-II in the late-endosome or the lysosome ( 30 ). The CLIP on MHC-II is driven out by interaction with another nonconventional MHC-II, called HLA-DM in humans and H2-M in mice ( 30 ). Then, MHC-II complexes can bind to antigen peptides and be presented on the cell surface ( 30 ). The expression of the peptide-MHC-II complex on the cell surface and its turnover by ubiquitination in DCs is essential for their ability to efficiently prime CD4 + T cells ( 31 , 32 ).
Molecular Basis of Antigen Cross-Presentation Pathway
Specific APCs are thought to take up extracellular antigens through endocytosis or phagocytosis and load peptides onto MHC-I for presentation to CD8 + T cells, a process called antigen cross-presentation ( 3 ). The extracellular antigen degradation pathway is mainly divided into the “vacuolar pathway”, through which the peptide is degraded in the endosome, and the “cytosolic pathway” which is responsible for the transport of degraded protein through SEC61 from the endosome to the cytosol ( 33 ) ( Figure 2 ).
Vacuolar Pathway of Antigen Cross-Presentation
In the vacuolar pathway, extracellular antigens are endocytosed by APCs and degraded into peptide fragments by proteases in the compartment. Cathepsin S plays a crucial role in antigen degradation in the endosomes of bone marrow-derived DCs (BMDCs) ( 34 ). It has been shown that cathepsin S plays a key role in priming CD8 + T cells to Influenza A virus (IAV) peptides loaded on MHC-I in the vacuolar pathway ( 34 ). In DCs, cathepsin S is also a crucial protease for MHC-II-dependent presentation to CD4 + T cells ( 18 , 35 ) whereas cathepsin L in the thymic cortical epithelium ( 35 ) and cathepsin F in macrophages likely correspond to proteases in the vacuolar pathway ( 36 ). The degraded peptide by cathepsins forms a complex with MHC-I in the endosome, and the peptide-MHC-I complex is transported to the cell surface. However, it is not clear whether cathepsins are required for antigen degradation in all lung APCs during pathogen infection.
Cytosolic Pathway of Antigen Cross-Presentation
In the cytosolic pathway, phagocytosed or endocytosed antigens are translocated from the endosomal compartments to the cytosol via Sec61 ( 33 ) and degraded to peptide fragments by the proteasome in the cytosol ( 37 , 38 ). Phagosomes and endosomes are mainly acidified via V-ATPase for degradation ( 39 ), which is regulated by Toll-like receptor (TLR) signals and other maturation signals ( 40 ), and restriction of antigen in these compartments by acidification is important for peptide degradation in the cytosolic pathway. DCs lacking the NADPH oxidase NOX2 show enhanced phagosomal acidification and increased antigen degradation, resulting in impaired antigen presentation ( 41 , 42 ). The recruitment of NOX2 to these compartments is prevented by deficiency of Rab27a, which causes acidification of phagosomes, limiting antigen degradation ( 43 ).
The cytosolic pathway is further categorized to two pathways; “ER-dependent pathway” and “Endosomal pathway”. The ER-dependent pathway is the most common route to ER for antigen peptide. Antigen peptides in the cytosol are transported into the ER mainly through transporter associated with antigen processing (TAP) and form peptide-MHC-I complexes in the ER. On the other hand, peptides degraded by the proteasome in the cytosol are transported back to the endosomes through TAP in the endosomal pathway. MHC-I molecules are recycled in the cells. MHC-I molecules in the endosome are transported from the plasma membrane through the Rab11 + recycling endosomes ( 44 ) and are also recruited from the ER or the ER-Golgi intermediate compartment (ERGIC) ( 3 , 45 ). Transported peptides are loaded on MHC-I by the peptide loading complex (PLC) the in the ER or the endosomes ( 46 ). PLC consists of TAP, oxidoreductase ERp57, MHC-I heterodimer, and calreticulin ( 46 ). PLC is recruited to phagosomes or endosomes via the Sec22b-ERGIC pathway ( 47 ). PLC is also recruited from the recycle endosomes after TLR activation ( 44 ). In contrast, the N terminal anchor residues of the peptides are trimmed by ER-resident N-aminopeptidases (ERAP1 and ERAP2 in humans, and ERAAP in mice). Insulin regulated aminopeptidase (IRAP), an aminopeptidase similar to ERAP, trims the peptide in the endosomes ( 48 , 49 ). These peptide trimming proteins are crucial for efficient antigen peptide binding to MHC-I and contribute to cross-presentation ( 50 – 52 ). Although cytosolic peptides shuttle into the ER through TAP1 in the cytosolic pathway, TAP1 blockade in DCs leads to antigen presentation by MHC-I translocation from ERGIC in a Sec22b-dependent manner rather than the Rab11 + recycle-endosome pathway ( 53 ).
DCs and MACs in the Lung
DCs in the lung consist of heterogeneous subsets that exert different functions ( 54 , 55 ). Lung DCs are largely divided into three major subsets and are broadly subdivided into plasmacytoid DCs (pDCs) and conventional DCs (cDCs). Murine cDCs express high levels of integrin CD11c and are further divided into CD103 + DC and CD11b + DCs. CD103 + DCs and CD11b + DCs are also referred to as cDC1s and cDC2s, respectively ( 55 – 58 ). Although CD11b and CD11c have been utilized for the separation of DC population, cDCs separation was proposed as two main subsets cDC1s and cDC2s based on the transcription factor expression ( 59 , 60 ). Interferon regulatory factor 8 (IRF8) and Batf3 drive the development of cDC1s which are separated as XCR1 + Cadm1 + CD172a − cDC1s ( 61 – 69 ). On the other hand, IRF4 drives the development of cDC2 which are separated as XCR1 − Cadm1 − CD172a + cDC1s ( 67 , 69 – 76 ). pDCs develop in the presence of transcription factor 4 (E2-2) and the Ets family transcription factor Spi-B ( 77 – 79 ). In the steady-state, cDC1s associate with airway rather than alveoli in the lung ( 80 , 81 ). cDC2s are located in the airway and lung parenchyma ( 82 – 84 ). Monocyte-derived DCs (moDCs) have been described as another DC population that accumulates in the lungs during inflammation and viral infection ( 85 – 87 ). MoDCs are also known as inflammatory DCs and monocyte derived cells ( 88 – 91 ). These DCs are subdivided based on the presence of surface markers and recent progress in the technology for single-cell RNA sequencing revealed that the cDC2s population in the lung is subdivided based on expression markers with functional differences, whereas pDCs and cDC1s are a unique population ( 92 – 94 ).
MACs in the lungs consist of two major populations: alveolar MACs (AMs) and interstitial MACs (IMs). AMs are located in the alveolar space of the lungs and are in close contact with the type I and II epithelial cells of the alveoli. AMs are the first line of defense against pathogens for host defense in the lung, with a higher engulfment capacity against antigens and pathogens ( 95 ). AMs produce cytokines such as TGFβ, IL6, and type I interferon during pathogen infection and inflammation ( 95 , 96 ). In addition, AMs play a central role in homeostasis and tissue remodeling. Pulmonary surfactant is a mixture of lipids and proteins secreted into the alveolar space by AT II cells. The surfactant is covered with an interface of alveolar epithelial cells in the lungs to reduce the physical tension during breathing. In addition, the engulfment of surfactant and cell debris by AMs is important for the clearance and maintenance of lung homeostasis. Accumulation of pulmonary surfactant in the absence of AMs causes the development of pulmonary alveolar proteinosis (PAP) ( 97 – 99 ). GM-CSF and TGF-β induce PPAR-γ, a crucial transcription factor for AM development ( 100 ). Interstitial macrophages (IMs) reside in the parenchyma between the microvascular endothelium and alveolar epithelium. However, compared with AMs, the role of IMs in lung homeostasis remains poorly understood. Like AMs, IMs engulf bacteria and foreign particles and secrete IL-1, IL-6, IL-10, and TNFα ( 101 – 104 ). IMs form a heterogeneous population that is further subdivided based on surface markers with distinct functions ( 103 , 105 ).
The lung is composed of a complex tissue structure that exchanges gas and is exposed to outer space. The combination of crosstalk between DCs and MACs effectively protects against inhaled pathogens by inducing acquired immunity ( Figure 3 ). cDC1s, cCD2s and IMs express high levels of MHC I/II with co-stimulatory molecules CD80 and CD86 ( 106 ). However, AMs express lower levels of MHC-II. Based on the expression of molecules for antigen presentation, it is revealed that each cells display antigen presentation capacity against specific infectious pathogens and allergic materials in the lungs.

Antigen presenting cells in the lung. The lungs are constantly exposed to environmental and infectious agents such as dust, viruses, fungi, and bacteria that invade the lungs upon breathing. The lungs are protected by various types of immune cells and epithelial cells. Lung DCs are largely divided into three major subsets and are broadly subdivided into pDCs, cDC1s and cDC2s. MACs in the lungs consist of two major populations: AMs and IMs. LECs consist of ATI and ATII cells in the alveoli, and the endothelial cells and other types of cells constituting the bronchial airway epithelium. Monocytes migrate to the lungs in response to inflammatory stimuli in a CCR2-dependent manner and these cells differentiate to moDCs or AMs. Small blood vessels allow oxygen to be extracted from the air into the blood, and carbon dioxide to be released from the blood into the air. The cells lining the inner surface of blood vessels are the pulmonary endothelial cells. These cells function as APCs that engulf pathogens through endocytosis or phagocytosis and present their peptides on major MHC-I or MHC-II protein complex to CD8 + or CD4 + T cells.
Antigen Presentation by pDCs
pDCs are professional cells that secrete type I IFN through the stimulation of innate immune receptors. It is widely accepted that the production of type I IFN by pDCs in the lungs is important for host defense against pathogens. An Aspergillus fumigatus infection model in the lung demonstrated that pDCs are essential for host defense and neutrophil effector activity ( 107 ). Antigen presentation by pDCs in the lungs is controversial during pathogen infection. Resting pDCs are weak antigen-presenting cells, but appear to be functionally specialized for their ability to capture and present viral antigens to CD4 + T cells in the presence of CpG DNA or virus stimulation ( 108 , 109 ). Transplantation of pDCs in an IAV infection model showed that pDCs infected with IAV promote antigen presentation to CD8 + T cells ( 110 ). In contrast, ablation of pDCs does not have a significant impact on the production of IAV-specific CD8 + T cells and viral clearance, indicating that pDCs have weak or no antigen cross-presentation capacity in vivo ( 111 ). Other groups have shown that pDCs in other peripheral tissues cooperate with cDC2s to promote their maturation and cross-presentation activity and induce antiviral CD8 + T cells, suggesting that pDCs indirectly induce antigen-specific CD8 + T cells ( 112 , 113 ).
Antigen Presentation by cDC2s
cDC2s are localized in the lungs under a steady-state condition, and a large number of cDC2s are accumulated in the lungs in response to inflammation induced by viral infection ( 114 ) or antigen immunization ( 115 ). IAV infection induces accumulation of cDC2s, and the depletion of these cells reduces the number of virus-specific CD8 + cells and mortality ( 85 – 87 ). These results indicate that accumulated cDC2s migrate to the lymph nodes and present antigens to CD8 + T cells. However, cDC1 analysis using Batf3 -deficient mice indicated that cDC2s have a weak cross-presentation capacity in vivo and support the proliferation of CD8 + T cells in the lung during IAV infection ( 116 ). Initial antigen-specific T cell differentiation is induced in the tissue-draining lymph nodes, and lung cDC2s are less migratory than cDC1s ( 117 ). During the inflammation, cDC2s in the lungs have shown to prime CD4 + Th2 cells but not CD8 + T cells responses ( 69 , 75 , 76 ). cDC2s also have shown to prime CD4 + Th17 cells response during Aspergillus fumigatus infection ( 74 ). T follicular helper (Tfh) cells are a subset of CD4 + T cells that promote antibody production during vaccination. cDC2s carry antigen into the lymph node where cDC2-dependent Tfh cells prime antibody-mediated protection from IAV challenge ( 67 ). cDC2s also locate in lymphoid organ, skin intestine and others organs as same with lung cDC2s, and cDC2s in the other organs efficiently promote the differentiation of CD4 + T cells into effector helper T cells during infection with Nippostrongylus brasiliensis , Aspergillus fumigatus or Citrobacterior rodentium ( 70 – 73 ). These results suggest that cDC2s are more specialized in polarizing CD4 + T helper cell responses and providing help to B cells, rather than in inducing CD8 + T cells activation.
cDC2s consist of heterogeneous subpopulations although it is unclear whether the same subpopulation of cDC2s induces both Th2 and Th17 cells ( 71 , 74 , 94 ). Single-cell RNA and cytometry by time-of-flight (CyTOF) analyses revealed that cDC2s consist of five distinct clusters. Ly-6C + CD301b – cDC2s promote Th17 differentiation, and CD200 + cDC2s induce the differentiation of Th2 but not Th17 cells ( 94 ). In addition, there are conflicting reports on how moDCs and CD11b + DCs interact with and regulate T cell responses ( 118 ). A recent report indicated that inflammatory cDC2s (inf-cDC2s) express the Fc receptor CD64 shared with moDCs and IRF8 shared with cDC1s and are infiltrated to present antigen to CD4 + and CD8 + T cells during respiratory virus infection ( 92 ). TNFR2 − cDC2 subpopulation drives moDCs maturation to generate T follicular helper (Tfh) cells in the lung ( 119 ).
Antigen Cross-Presentation by cDC1s
Many studies have shown the importance of cDC1s in the initiation of antiviral T cell response following influenza infection. Particular subsets of cDC1s, such as CD8α + and CD103 + cDC1s, play specific roles in naïve T cell activation and differentiation ( 10 , 120 – 122 ). CD8α + cDC1s in the spleen and lymphoid organs are known as the cross-presenting subset ( 123 – 125 ). CD103 + cDC1s are migratory DCs that cross-present antigens in peripheral tissues, including the lungs ( 126 , 127 ). Both CD103 + cDC1s in the lungs and CD8α + cDC1s in lymph nodes share the expression of various genes, including transcription factors IRF8, BATF3, and ID2, and both of these DC subtypes are developed in the presence of Flt3 ( 128 ).
cDC1s directly present antigen to naive CD4 + T cells ( 129 ) and cDC1s could prime Th2 and Th17 differentiation by producing IL4, IL12, IL13 and IL17 induction during allergic airway inflammation ( 130 , 131 ). A mouse model of invasive pulmonary aspergillosis infection showed cDC1s induces Th17 response by producing IL-2 in the lung ( 132 ). Other reports postulate that cDC1s promote airway tolerance by the induction of FoxP3 + T reg s in antigen induced airway inflammation ( 133 ) or by inducing IL-10 without T reg -induction ( 134 ). Although cDC1s can present antigens and stimulate CD4 + T cells, they are well known for their ability to cross-present antigens to CD8 + T cells ( 127 , 132 ). Lung cDC1s preserve viral antigens in their endocytic compartments and control the induction of virus-specific CD8 + T cells through antigen cross-presentation ( 116 , 135 ). Lung cDC1s migrate to mediastinal LNs after viral infection, where they directly present antigens to naïve CD8 + T cells or transfer captured antigens to CD8α + cDC1s, which present antigens and activate naïve CD8 + T cells ( 86 , 136 , 137 ). In addition to cDC1s, cDC2s have the potential to migrate to mediastinal LNs (MLNs) ( 117 ), however, cDC2s do not present antigens efficiently in the MLNs ( 138 ). The cytotoxic activity of CD8 + T cells plays a critical role in viral clearance in the lungs. Initial virus-specific CD8 + T cells in the LNs are induced by cDC1s migrating from the infected lung, and the virus-specific CD8 + T cells then traffic back to the infected lung to mediate their effector function ( 10 , 11 , 139 ).
Antigen Presentation by moDCs
Chemokine receptor CCR2- and Ly6C-expressing inflammatory monocytes infiltrate into the lung during pathogen infection including Aspergillus fumigatus ( 140 ) and IAV ( 141 ), and differentiate rapidly into moDCs. MoDCs in other organs are also capable of presenting antigen and priming to CD4 + T cells ( 142 , 143 ) and CD8 + T cells ( 88 ). However, the precise function of moDCs to regulate T cells response in lung is controversial. CCR2-deficient mice impair moDCs recruitment and exhibit reduction of effecter CD8 + T cell response in the lung after IAV infection ( 85 ). moDCs depletion by CD11c-cre- Irf4 f/f mice reduces CD8 + memory precursor cells and T RM cells during IAV infection ( 144 ). MoDCs in the lung prime IFN-γ-producing antigen-specific CD4 + T cells in pulmonary aspergillosis ( 140 ). MoDCs also promote Th1 and Th17 cell polarization through antigen presentation during allogeneic responses ( 118 ) and induce Th2 type CD4 + cells during house dust mite allergy ( 145 ). Report using CD26 as a maker for separation of moDCs indicated that moDCs have poor capacity to migrate to lymph node and prime CD4 + T cells and CD8 + T cells ( 92 , 117 , 146 ).
Antigen Presentation by Macrophages
AMs develop during embryogenesis, and then predominantly maintain their populations by self-renewal ( 147 – 149 ) and are specialized in the removal and recycling of surfactant molecules. Although AMs are the most abundant immune cells in the lungs and have been suggested to play a functional role in antigen presentation during tuberculosis and Cryptococcus neoformans infection in humans ( 150 , 151 ), supportive evidence for antigen presentation by AMs has not been reported in mice. Certain IM subsets have been contributed to lung immune homeostasis by spontaneously producing the immunosuppressive cytokine IL-10 and preventing the development of aberrant type 2 allergic responses against inhaled allergens ( 101 , 104 ). IMs are separated by a distinct subpopulation based on the surface expression pattern ( 103 , 152 ) and single-cell RNA sequencing ( 153 , 154 ), some of which express antigen-presenting genes and may mediate antigen presentation to CD4 + T cells in the lungs. Accumulated Ly-6C + monocytes develop to exudative macrophages (exMACs) during Cryptococcus ( 155 ), Streptococcus ( 156 ) and IAV infection ( 157 ). ExMACs produce high levels of TNF-α and NOS2 and stimulate the proliferation of memory CD4 + T cells ( 157 ).
Antigen Presentation by Monocytes
Two types of monocytes have been identified with different phenotypes and functions: Ly6C + classical monocytes and Ly6C − non-classical monocytes. Ly6C + monocytes constitutively enter to lung tissues in the steady state and a large number of these cells migrate to the lungs in response to inflammatory stimuli in a CCR2-dependent manner ( 158 , 159 ). Ly6C + monocytes develop to moDCs, IMs, exMACs or monocyte-derived AMs in the lungs during inflammatory stimulation, but in the steady state, monocytes continuously migrate to non-lymphoid organs including lung without differentiating into other types of cells and may exit lung via the lymphatics or undergo local apoptosis and cleared ( 160 ). Ly6C + monocytes have been shown to produce large amounts of IL-1, IL-6, and TNFα, and have an ability to drive adaptive immune responses through antigen presentation ( 160 ). Ly6C + monocytes in other tissues reported that these cells have an ability to present antigen to both CD4 + and CD8 + cells. Ly6C + monocytes regulate early host response to Aspergillus lung infection by taking up conidia and trafficking them into the draining LN to prime CD4 + T cells ( 140 ). Cross-presentation by Ly6C + inflammatory monocytes in lymphoid organs has been reported in the presence of TLR agonists, especially TLR7 ( 161 ). Once recruited into the lungs, Ly6C + monocytes further differentiate into moDCs and monocyte-derived AMs. Recent evidence have shown that CCR2-deficient mice, which are defective in monocyte trafficking to the lung, exhibit decreased number of virus-specific lung resident memory CD8 + (T RM ) cells by the antigen presentation on monocytes ( 162 ).
Antigen Presentation by Epithelial and Endothelial Cells
As lung epithelial cells directly interact with the external environment, these cells are thought to be critical regulators of barrier immunity ( 163 , 164 ). The alveoli are composed of two distinct lung epithelial cell types: AT I cells, which are thin and cover approximately 95% of the internal surface of the lung, and AT II cells, which are cuboidal secreting cells located between type I cells ( 165 ). AT I cells are specialized in gas exchange and alveolar fluid regulation, whereas type II cells secrete surfactants and constitute the progenitor cells of the epithelium ( 166 ). There is increasing evidence that epithelial cells in the lung contribute to adaptive immune responses in the lungs. AT II cells express MHCII and present antigen. In vitro co-culture experiments AT II cells with antigen specific hybridoma suggested that AT II cells activate CD4 + cells to induce IFNγ in the presence of peptide antigen, and deletion of MHC-II on AT II cells results in a modest worsening of respiratory virus disease following influenza and Sendai virus infections ( 167 ). Surfactant Protein C (SPC) low MHC-II high AT II cells function as APCs to induce CD4 + T RM cells ( 7 ). Antigen presenting AT II cells primes naïve CD4 + T cells in vitro and induce regulatory T (T reg ) cells ( 168 ); however, it is unclear whether AT II cells prime naïve CD4 + T cells in vivo ( 169 ). In addition to CD4 + T cells activation, barrier epithelial cells recruit and maintain CD8 + T RM cells near the sites of antigen encounter and reactivate them in the tissues via local antigen presentation ( 12 , 170 ).
Small blood vessels, known as capillaries, come in close contact with the alveoli, allowing oxygen to be extracted from the air into the blood, and carbon dioxide to be released from the blood into the air. The cells lining the inner surface of these capillaries are known as the pulmonary endothelial cells ( 171 ). Lung endothelial cells cross-present malaria antigen to antigen specific reporter cells in vitro and a mouse model of malaria infection by Plasmodium berghi ANKA (PbA) induces IFNγ positive CD8 + T cell. These results demonstrate that lung endothelial cells cross-present malaria antigen to CD8 + T cells, although it is unclear whether these cells activate naive CD8 + T cell in vivo ( 172 ).
Perspective and Conclusion
Lungs are protected by various types of APCs that stimulate antigen-specific CD4 + and CD8 + T cells against infectious pathogens. cDC1s and cDC2s work as professional APCs in the lung. Sub-population of cDCs has been investigated by deep separation using single RNA sequence and CyTOF technology and have shown to process and present antigen. In addition, there has been increasing evidence for antigen presentation by resident APCs such as epithelial cells, epithelial cells in the lungs. The relation of pathogen and inflammation model to APCs was shown in Table 1 . Although MACs express MHC and costimulatory molecules with higher engulfment capacity, the role of MACs in the lung as APCs is still unclear.
Lung APCs and their roles in T cell responses.
To initially prime antigen-specific T cells, antigen-captured DCs and migratory APCs need to traffic to lung-draining LNs where they encounter naïve T cells to select antigen-specific T cells. Following a program of proliferation and differentiation of T cells in LNs, antigen-specific effector or memory T cells migrate back to the infected lung to mediate their effector function ( 10 , 11 ). At the same time, antigen-specific effector or memory T cells are reactivated by APCs, including monocytes, epithelial cells and endothelial cells in the lungs, with support of cytokine production and the local microenvironment ( 12 ). Among the antigen specific memory type cells, CD4 + and CD8 + T RM cells in the lung provide protection against pathogen infection and retain for long time period in the peripheral tissue. Pulmonary antigen encounter is necessary for the establishment of T RM during IAV infection in the lung ( 173 ), and antigen presentation by DCs with cytokines such as TGF-β and IL15 is shown to be important for T RM development in the lung ( 174 – 176 ). Various types of APCs in the lungs contribute to pathogen clearance against viruses, fungi, and bacteria; therefore, APCs perform their function depending on the pathogen infection, and further studies are needed to clarify the role of individual APCs in the lungs.
Author Contributions
TakK and MI wrote the manuscript. TarK edited and supervised the manuscript. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank Chihiro Suzuki for secretarial assistance. This work was supported by JSPS KAKENHI Grant-in-Aid for Scientific Research B (20H03468) and C (19K07608), Grant-in-Aid for Early-Career Scientists (21K14817) and the Takeda Science Foundation. We would like to thank Editage ( www.editage.com ) for English language editing.
AI Generator

Antigens are substances that trigger an immune response in the body. These molecules are often proteins or polysaccharides found on the surface of pathogens like bacteria , viruses, and fungi. When the immune system detects an antigen, it produces antibodies to neutralize or destroy the foreign invader. Antigens can also be toxins or other foreign substances that induce an immune response . They play a crucial role in the body’s ability to recognize and fight off infections, making them essential components of the immune system. Understanding antigens is key to developing vaccines and therapies for various diseases.
What is an Antigen?
An antigen is any substance that provokes an immune response in the body. These substances are usually proteins or polysaccharides found on the surface of pathogens such as bacteria, viruses, or fungi. When the immune system recognizes an antigen as foreign, it produces specific antibodies to target and neutralize the invader. Antigens can also include toxins or other foreign substances that elicit an immune reaction.
Examples of Antigens in Immune Response, Blood, and the Human Body
- Bacterial Toxins – Substances produced by bacteria, such as the diphtheria toxin, that trigger an immune response.
- Viral Proteins – Surface proteins of viruses like the spike protein of the SARS-CoV-2 virus, which causes COVID-19.
- Pollen – Common allergen from plants that can cause allergic reactions in sensitive individuals.
- Dust Mites – Proteins from dust mite feces that can induce allergic responses.
- Blood Group Antigens – Surface molecules on red blood cells that determine blood types, such as ABO and Rh factors.
- Autoantigens – Normal body proteins mistakenly targeted by the immune system in autoimmune diseases, like insulin in type 1 diabetes.
- Tumor Antigens – Abnormal proteins expressed on cancer cells that the immune system can recognize and attack.
- Food Proteins – Proteins in foods like peanuts or shellfish that can cause allergic reactions.
- Vaccines – Inactivated or attenuated pathogens, or parts of pathogens, introduced to stimulate an immune response without causing disease.
- Parasite Antigens – Proteins on the surface of parasites, such as those from malaria-causing Plasmodium species, that trigger an immune response.
Types of Antigens

Based on Origin
- Origin: External environment
- Examples: Bacterial toxins, viruses, pollen
- Role: Enter the body through inhalation, ingestion, or injection and are processed by antigen-presenting cells (APCs) like dendritic cells, macrophages, and B cells.
- Origin: Inside the body’s cells
- Examples: Viral proteins produced inside infected cells, abnormal proteins in cancer cells
- Role: Presented on the cell surface by MHC class I molecules and recognized by cytotoxic T cells.
- Origin: Normal body tissues
- Examples: Myelin basic protein in multiple sclerosis, insulin in type 1 diabetes
- Role: Typically ignored by the immune system, but in autoimmune diseases, they are mistakenly targeted by the immune response.
Based on Function
- Characteristics: Can trigger an immune response without assistance from T-helper cells
- Examples: Bacterial polysaccharides, lipopolysaccharides
- Role: Directly stimulate B cells to produce antibodies.
- Characteristics: Require T-helper cells to provoke an immune response
- Examples: Proteins from pathogens, vaccines
- Role: B cells process and present these antigens to T-helper cells, which then aid in B cell activation and antibody production.
Based on Immunogenicity
- Characteristics: Can induce a full immune response on their own
- Examples: Whole pathogens like bacteria, viruses
- Role: Possess both immunogenicity (ability to provoke an immune response) and antigenicity (ability to specifically bind to antibodies or immune cells).
- Characteristics: Cannot induce an immune response by themselves
- Examples: Small molecules like penicillin, urushiol (poison ivy toxin)
- Role: Become immunogenic only when attached to larger carrier molecules, forming a complete antigen.
Antigen-Presenting Cells (APCs)
Antigen-presenting cells (APCs) are a crucial component of the immune system. They capture, process, and present antigens to T cells, initiating an immune response. APCs display fragments of antigens on their surface using molecules called major histocompatibility complex (MHC) proteins. This presentation allows T cells to recognize and respond to the antigens, either by activating other immune cells or by directly attacking infected cells.
Types of Antigen-Presenting Cells
- Location : Found in tissues that are in contact with the external environment, such as the skin (Langerhans cells), and the inner lining of the nose, lungs, stomach, and intestines.
- Function : Capture antigens, migrate to lymph nodes, and present them to T cells. They are the most effective APCs and are essential for initiating T cell responses.
- Location : Present in almost all tissues, particularly abundant in the liver (Kupffer cells), lungs (alveolar macrophages), and lymph nodes.
- Function : Engulf and digest pathogens and debris. After processing antigens, they present them to T cells, especially during the later stages of an immune response.
- Location : Found in the blood, lymph nodes, and spleen.
- Function : Bind specific antigens through their B cell receptors (BCR), process the antigens, and present them to helper T cells. This interaction is crucial for B cell activation and subsequent antibody production.
Antigen Functions
1. immune system activation.
Antigens trigger the activation of the immune system. When antigens are detected, the body responds by producing antibodies and activating various immune cells to combat the invading pathogens.
2. Specificity
Antigens determine the specificity of the immune response. Each antigen has a unique molecular structure that is recognized by specific antibodies or immune cells, ensuring that the immune response targets the correct pathogen.
3. Memory Formation
Antigens are essential for the formation of immunological memory. Once the immune system encounters an antigen, it “remembers” it, leading to a quicker and more efficient response if the same antigen is encountered again in the future.
4. Pathogen Identification
Antigens help the immune system identify and differentiate between self and non-self molecules. This is crucial in distinguishing between the body’s own cells and harmful pathogens like bacteria, viruses, and fungi.
5. Vaccination Response
Vaccines contain antigens that mimic specific pathogens. When administered, these antigens stimulate the immune system to produce a protective response without causing disease, thereby providing immunity against future infections by the actual pathogen.
6. Activation of T-Cells
Antigens are presented by antigen-presenting cells (APCs) to T-cells, which are critical components of the adaptive immune system. This interaction leads to the activation and proliferation of T-cells, which then assist in eliminating the pathogen.
7. B-Cell Activation
Antigens bind to B-cell receptors on the surface of B-cells. This binding, along with signals from helper T-cells, activates B-cells to proliferate and differentiate into plasma cells that produce antibodies specific to the antigen.
8. Allergy Development
In some cases, antigens can trigger allergic reactions. These antigens, known as allergens, cause the immune system to overreact, leading to symptoms like inflammation, itching, and respiratory issues.
9. Autoimmune Responses
In autoimmune diseases, the immune system mistakenly targets self-antigens, which are the body’s own molecules. This leads to the immune system attacking healthy tissues, causing conditions like rheumatoid arthritis and lupus.
Where Are Antigens Found?
Antigens are substances that trigger an immune response in the body. They can be found in various locations and forms, including:
1. On the Surface of Pathogens
- Bacteria : Antigens are present on the cell walls, membranes, and flagella.
- Viruses : Viral antigens are found on the protein coat or envelope surrounding the viral genome.
2. On Infected Cells
- Virus-Infected Cells : When a virus infects a cell, viral proteins (antigens) are displayed on the cell’s surface.
- Bacteria-Infected Cells : Similarly, cells infected by bacteria may present bacterial antigens on their surfaces.
3. In Vaccines
- Live Attenuated Vaccines : Contain weakened pathogens with surface antigens.
- Inactivated Vaccines : Contain killed pathogens or specific parts of pathogens (like proteins) that act as antigens.
- Subunit Vaccines : Include only the antigens that best stimulate the immune system.
4. On Transplanted Organs and Tissues
- Donor Tissues : Organs or tissues from donors have antigens that may be recognized as foreign by the recipient’s immune system, potentially leading to rejection.
5. In Blood and Body Fluids
- Blood Types : Blood group antigens are found on the surface of red blood cells (A, B, AB, and O antigens).
- Allergens : Certain proteins in food, pollen, and other substances can act as antigens, causing allergic reactions.
6. On Cancer Cells
- Tumor Antigens : Cancer cells often express unique antigens or overexpress normal proteins, which can be recognized by the immune system.
How Do Antigens Enter the Body?
Antigens are substances that can trigger an immune response. They are typically foreign molecules such as bacteria, viruses, fungi, and toxins. Understanding how antigens enter the body is crucial for comprehending how our immune system detects and responds to potential threats. Here are the primary ways antigens can enter the body:
1. Respiratory Tract
The respiratory tract is one of the most common entry points for antigens. Airborne pathogens like bacteria, viruses, and allergens can enter the body through inhalation. Once inside, these antigens can cause respiratory infections and trigger immune responses.
- Viruses: Influenza, COVID-19
- Bacteria: Streptococcus pneumoniae, Mycobacterium tuberculosis
- Allergens: Pollen, dust mites
2. Gastrointestinal Tract
The gastrointestinal (GI) tract allows antigens to enter the body through ingestion. Contaminated food and water can introduce bacteria, viruses, and parasites into the digestive system.
- Bacteria: Salmonella, Escherichia coli
- Viruses: Norovirus, Hepatitis A
- Parasites: Giardia, Entamoeba histolytica
The skin serves as a physical barrier, but antigens can still enter through cuts, abrasions, or insect bites. Direct contact with contaminated surfaces or objects can also introduce pathogens through the skin.
- Bacteria: Staphylococcus aureus (through cuts)
- Viruses: Rabies virus (through animal bites)
- Parasites: Plasmodium (through mosquito bites)
4. Urogenital Tract
Sexually transmitted infections (STIs) are common ways for antigens to enter the body through the urogenital tract. Pathogens can be transmitted during sexual contact.
- Bacteria: Neisseria gonorrhoeae, Chlamydia trachomatis
- Viruses: Human Immunodeficiency Virus (HIV), Herpes Simplex Virus (HSV)
5. Bloodstream
Antigens can directly enter the bloodstream through intravenous routes. This can occur via contaminated needles, transfusions, or insect vectors.
- Viruses: Hepatitis B and C (through contaminated needles)
- Bacteria: Borrelia burgdorferi (through tick bites causing Lyme disease)
- Parasites: Trypanosoma (through insect vectors causing Chagas disease)
6. Mucous Membranes
Mucous membranes line various cavities in the body and can serve as entry points for antigens. These include the eyes, nose, mouth, and genitals.
- Viruses: Adenovirus (through the eyes), Human Papillomavirus (HPV) (through the genitals)
- Bacteria: Neisseria meningitidis (through the nose)
What happens When an Antigen Enters Your Body?
When an antigen enters your body, the immune system quickly detects it as a foreign invader. Antigen-presenting cells (APCs) like dendritic cells, macrophages, and B cells engulf and process the antigen, displaying its fragments on their surface bound to major histocompatibility complex (MHC) molecules. This presentation activates helper T cells, which in turn stimulate B cells to produce specific antibodies and cytotoxic T cells to attack infected cells. The antibodies neutralize or mark the antigen for destruction, while cytotoxic T cells directly kill infected cells. This coordinated response aims to eliminate the antigen and protect the body from harm.
What is the Strongest Antigen?
The strongest antigen is generally considered to be the protein antigen , due to its complex and diverse structure, which can elicit a robust immune response. Proteins are made up of long chains of amino acids folded into specific shapes, providing numerous unique sites for immune cells to recognize and bind. This makes protein antigens highly effective at triggering both the humoral (antibody-mediated) and cellular immune responses. Examples of strong protein antigens include those found on the surfaces of pathogens like the hemagglutinin protein of the influenza virus and the spike protein of the SARS-CoV-2 virus, both of which are highly immunogenic and essential targets for vaccines.
Self Antigen
Self-antigens are normal proteins or molecules present within the body that the immune system typically recognizes as “self” and does not attack. These antigens are crucial for maintaining immune tolerance, preventing the immune system from targeting the body’s own tissues. In autoimmune diseases, this tolerance breaks down, leading the immune system to mistakenly identify self-antigens as foreign. This results in an immune response against the body’s own cells and tissues, causing inflammation and damage. Examples of self-antigens involved in autoimmune diseases include myelin basic protein in multiple sclerosis and insulin in type 1 diabetes. Understanding self-antigens is essential for developing treatments that restore immune tolerance and prevent autoimmunity.
Properties of Antigens
1. immunogenicity.
Immunogenicity refers to the ability of an antigen to provoke an immune response. An effective antigen must be recognized as foreign by the immune system and must be able to stimulate the production of specific antibodies or activate specific immune cells.
2. Antigenicity
Antigenicity is the capacity of an antigen to bind specifically to the products of the immune response, such as antibodies or T-cell receptors. An antigen’s antigenicity determines how well it can be identified and targeted by the immune system.
3. Molecular Size
Larger molecules are generally more immunogenic than smaller ones. Typically, molecules with a molecular weight of over 10,000 daltons are considered good antigens, as their size and complexity provide multiple epitopes for immune recognition.
4. Chemical Complexity
Antigens with a more complex chemical structure tend to be more immunogenic. Proteins, with their diverse amino acid sequences and complex three-dimensional structures, are usually stronger antigens compared to simpler molecules like lipids or polysaccharides.
5. Foreignness
The degree of foreignness to the host organism influences an antigen’s immunogenicity. The more different an antigen is from the host’s own molecules, the stronger the immune response it will elicit. This is because the immune system has evolved to distinguish self from non-self molecules.
6. Epitope Density
Epitopes are specific parts of an antigen that are recognized by the immune system. Antigens with a high density of epitopes are more likely to be immunogenic because they provide multiple binding sites for antibodies or T-cell receptors.
7. Degradability
Antigens that can be easily processed and presented by antigen-presenting cells (APCs) are more likely to elicit an immune response. Degradability allows antigens to be broken down into peptides that can be displayed on the surface of APCs for recognition by T-cells.
8. Route of Entry
The route by which an antigen enters the body can affect its immunogenicity. Different routes, such as intramuscular, subcutaneous, oral, or respiratory, can influence the type and magnitude of the immune response.
The amount of antigen introduced into the body can impact the immune response. Both very low and very high doses of an antigen may result in weak immune responses, whereas an optimal intermediate dose tends to induce a stronger response.
What are antigens?
Antigens are molecules that trigger an immune response, often found on the surface of pathogens.
How do antigens work?
Antigens alert the immune system to the presence of foreign substances, prompting an immune response.
What are examples of antigens?
Examples include proteins, polysaccharides, lipids, and nucleic acids found on bacteria, viruses, and other pathogens.
What is the role of antigens in vaccines?
Vaccines use antigens to stimulate the immune system, creating immunity without causing disease.
How are antigens detected?
Antigens are detected through immune assays like ELISA and rapid tests, identifying specific immune responses.
What is an antigenic determinant?
An antigenic determinant, or epitope, is the specific part of an antigen recognized by the immune system.
How do antigens differ from antibodies?
Antigens trigger immune responses, while antibodies are proteins that specifically bind to antigens to neutralize them.
What are self and non-self antigens?
Self-antigens are body’s own molecules, while non-self antigens are foreign, prompting an immune response.
What is an antigen-presenting cell?
Antigen-presenting cells process and present antigens to T-cells, initiating an immune response.
Can antigens cause allergies?
Yes, allergens are antigens that cause allergic reactions by triggering an overactive immune response.
Text prompt
- Instructive
- Professional
10 Examples of Public speaking
20 Examples of Gas lighting
HOOKIPA Pharma Announces Positive Clinical Data to be Presented at the American Society for Clinical Oncology 2024 Annual Meeting
May 23, 2024 17:01 ET | Source: HOOKIPA Pharma Inc. HOOKIPA Pharma Inc.
- HOOKIPA to present an oral abstract at the American Society for Clinical Oncology (ASCO) 2024 Annual Meeting on June 4
- Updated data of HB-200 plus pembrolizumab demonstrate a favorable safety profile and promising clinical activity
- In a subset of patients with PD-L1 combined positive score (CPS) of 20 or higher, data showed confirmed objective response rate (ORR) of 53%, complete response (CR) rate of 18%, and disease control rate (DCR) of 82%
- Company will also present promising preliminary progression-free survival and overall survival data for patients with CPS ≥20 on June 4
NEW YORK and VIENNA, May 23, 2024 (GLOBE NEWSWIRE) -- HOOKIPA Pharma Inc. (NASDAQ: HOOK, ‘HOOKIPA’), a company developing a new class of immunotherapeutics based on its proprietary arenavirus platform, today announced positive updated results from its Phase 1/2 clinical trial of HB-200 for the treatment of human papillomavirus 16 positive (HPV16+) head and neck cancers. The data were published in the Company’s abstract for the ASCO 2024 Annual Meeting and support the Company’s pivotal Phase 2/3 trial design for HB-200 in combination with pembrolizumab in the first line setting.
The abstract reported data as of January 12, 2024, and included 42 patients treated with HB-200 plus pembrolizumab. The treatment was generally well tolerated with a low rate of treatment-related discontinuation and no treatment-related deaths.
Among a subpopulation of 17 evaluable patients with CPS of 20 or higher, the updated data showed confirmed ORR of 53 percent, CR rate of 18 percent, and DCR of 82 percent. This subpopulation is representative of patients eligible for the Company’s pivotal Phase 2/3 trial, which will begin enrolling patients in the fourth quarter of 2024.
Additional data will be presented in the Head and Neck Oral Abstract Session at the ASCO 2024 Annual Meeting on June 4, at 11:09 a.m. CDT. During the presentation, preliminary progression-free survival and overall survival data will be shared for the first time.
“We are happy to provide an update on our clinical data and showcase the meaningful outcomes we are helping to drive for patients,” said Joern Aldag, Chief Executive Officer of HOOKIPA. “The data exhibit strong evidence that has helped inform our pivotal Phase 2/3 trial design, which will begin enrolling patients later this year. This update gives us conviction that we are on the right path to achieve our goals and help provide a new targeted therapeutic option for patients battling HPV16+ head and neck cancer.”
Results: HB-200 in combination with pembrolizumab: The abstract presented data as of January 12, 2024, and included 42 first line patients with HPV16+, PD-L1 positive, recurrent or metastatic head and neck squamous cell carcinoma. The updated data continue to demonstrate a favorable safety profile of HB-200 in combination with pembrolizumab and promising clinical activity as a first line treatment. Median follow-up time was 5.6 months.
HB-200 + pembrolizumab were generally well tolerated. Grade ≥3 treatment-related adverse events (TRAEs) were reported in 6 (14%) patients, serious TRAEs were reported in 3 (7%) patients, and TRAEs leading to treatment discontinuation were reported in 2 (5%) patients. No treatment-related deaths were reported.
Among 35 evaluable patients—those with ≥ 1 post-baseline tumor response assessment—3 confirmed complete responses, 9 confirmed partial responses, and 3 unconfirmed partial responses were observed. Notably, among patients with PD-L1 CPS ≥20 (N=17), ORR, based on confirmed responses, was 53%, CR rate was 18%, and DCR was 82%.
Additional Abstracts at ASCO 2024 Annual Meeting: HB-200 plus chemotherapy (neoadjuvant setting): In addition, data from an Investigator Initiated Trial (IIT), led by Dr. Ari Rosenberg of the University of Chicago Department of Medicine, will be presented on June 3, in the Head and Neck Rapid Oral Abstract Session. The study concluded that neoadjuvant HB-200 plus chemotherapy for the treatment of patients with non-metastatic HPV16+ oropharyngeal cancers (OPC) is safe and feasible, with early efficacy signal in this setting warranting further study.
Twenty-one patients with HPV16+ OPC were enrolled and treated across multiple cohorts and dose levels. All patients completed neoadjuvant HB-200/chemotherapy and response-stratified locoregional treatment. Deep responses following HB-200/chemotherapy were observed in 17/21 (81%) patients, and in 14/15 (93%) patients treated with higher dose levels 1 or 2. All three patients who underwent transoral robotic surgery (TORS) had no viable tumor at time of surgery. Two patients (9%) had persistent disease following chemoradiotherapy and underwent salvage surgery with no evidence of disease at last follow-up. ctHPV-DNA and HPV16-specific T-cell response data will be presented at the meeting.
Enrollment to the subsequent randomized phase II part is ongoing. Details of the rapid oral presentation are included below.
HB-700 preclinical data: HB-700 is an investigational arenaviral immunotherapy designed to treat KRAS-mutated lung, colorectal, pancreatic and other cancers. HB-700 is a replicating 2-vector therapy that targets the most common KRAS mutations (G12D, G12V, G12R, G12C and G13D) and has the potential to benefit a broader patient population than single mutation inhibitors.
A transgene cassette consisting of peptide stretches including KRAS mutations G12D, G12V, G12C, G12R and G13D was generated by in silico aided antigen design. KRAS mutation specific CD8+ T cell expansion was evaluated in HLA transgenic mice treated with HB-700 and functionality of induced CD8+ T cells was evaluated by assessing CD8+ T cell mediated killing of mutant KRAS peptide loaded target cells in vivo.
All treatment regimens were well tolerated, and no mortalities or major adverse events were observed. The results indicate efficient induction of KRAS mutation specific T cell responses in HLA transgenic mice. Expanded CD8+ T cells were capable of killing cells loaded with KRAS mutation specific peptides in vivo indicating functionality of the induced T cell responses. No specific cytotoxicity towards target cells pulsed with KRAS wild type peptides was observed in any of the groups. HOOKIPA’s HB-700 investigational product candidate differs from KRAS inhibitors and has a wide range of combinability options including with small-molecule inhibitors. Based on these results, initiation of a Phase I study for the treatment of KRAS mutated cancers is planned.
Abstract details: ASCO 2024 Annual Meeting
HB-200: Title: HB-200 arenavirus-based immunotherapy plus pembrolizumab as first-line treatment of patients with recurrent/metastatic HPV16-positive head and neck cancer: Updated results Presenter: Dr. Alan L. Ho, Head and Neck Oncologist at Memorial Sloan Kettering Cancer Center and a trial investigator Abstract Type: Oral abstract Session Name: Head and Neck Cancer Session Date and Time: June 4, 2024; 9:45 AM-12:45 PM CDT Abstract Number: 6005
Investigator Initiated Trial: Title: Neoadjuvant HPV16-specific arenavirus-based immunotherapy HB-200 plus chemotherapy followed by response-stratified de-intensification in HPV16+ oropharyngeal cancer: TARGET-HPV Presenter: Dr. Ari Rosenberg, Principal Investigator, TARGET-HPV Trial, University of Chicago Medicine Abstract Type: Rapid oral abstract Session Name: Head and Neck Cancer Session Date and Time: June 3, 2024; 8:00 AM-9:30 AM CDT Abstract Number: 6017 Trial Sponsor: UChicago Medicine
HB-700 Title: Development of an arenavirus-based immunotherapy for treatment of KRAS mutant cancer Abstract Type: Abstract only Session Date: May 23, 2024 Abstract Number: e14672
About HB-200 HB-200 is HOOKIPA’s lead oncology candidate engineered with the company’s proprietary replicating arenaviral vector platform. It comprises two single-vector compounds with arenaviral backbones based on lymphocytic choriomeningitis virus (LCMV) and pichinde virus (PICV). Both express the same transgene encoding an E7E6 fusion protein derived from HPV16. HB-200 is an alternating 2-vector immunotherapy designed to further focus the immune response against the encoded antigen.
HB-200 in combination with pembrolizumab received Fast Track Designation from the U.S. Food and Drug Administration and PRIME designation from the European Medicines Agency for the treatment of first-line HPV16+ recurrent/metastatic oropharyngeal squamous cell carcinoma. These designations are supported by preliminary clinical evidence from the Phase 1/2, open-label, clinical trial (NCT04180215) evaluating safety, T cell response, and efficacy based on objective response rate (ORR) and disease control rate (DCR) as defined by RECIST 1.1. and iRECIST.
About HB-700 HB-700 is an investigational arenaviral immunotherapy designed to treat KRAS-mutated lung, colorectal, pancreatic and other cancers. HB-700 is a replicating 2-vector therapy that targets the most common KRAS mutations (G12D, G12V, G12R, G12C and G13D) and has the potential to benefit a broader patient population than single mutation inhibitors.
About HOOKIPA HOOKIPA Pharma Inc. (NASDAQ: HOOK) is a clinical-stage biopharmaceutical company focused on developing novel immunotherapies, based on its proprietary arenavirus platform, which are designed to mobilize and amplify targeted T cells and thereby fight or prevent serious disease. HOOKIPA’s replicating and non-replicating technologies are engineered to induce robust and durable antigen-specific CD8+ T cell responses and pathogen-neutralizing antibodies. HOOKIPA’s pipeline includes its wholly owned investigational arenaviral immunotherapies targeting Human Papillomavirus 16-positive cancers, KRAS-mutated cancers, and other undisclosed programs. In addition, HOOKIPA aims to develop functional cures of HBV and HIV in collaboration with Gilead.
Find out more about HOOKIPA online at www.hookipapharma.com .
Forward Looking Statements Certain statements set forth in this press release constitute “forward-looking” statements within the meaning of the Private Securities Litigation Reform Act of 1995, as amended. Forward-looking statements can be identified by terms such as “anticipates”, “believes,” “expects,” “plans,” “potential,” “will,” “would” or similar expressions and the negative of those terms. Forward-looking statements in this press release include HOOKIPA’s statements regarding the potential of its product candidates to positively impact quality of life and alter the course of disease in the patients it seeks to treat, HOOKIPA’s plans, strategies, expectations and anticipated milestones for its preclinical and clinical programs, including the timing of initiating clinical trials and patient enrollment, the availability and timing of results from preclinical studies and clinical trials, the timing of regulatory filings, the expected safety profile of HOOKIPA’s product candidates, and the probability of successfully developing and receiving regulatory approval for its product candidates. Such forward-looking statements involve substantial risks and uncertainties that could cause HOOKIPA’s research and clinical development programs, future results, performance or achievements to differ significantly from those expressed or implied by the forward-looking statements. Such risks and uncertainties include, among others, the uncertainties inherent in the drug development process, including HOOKIPA’s programs’ early stage of development, the process of designing and conducting preclinical and clinical trials, plans and timelines for the preclinical and clinical development of its product candidates, including the therapeutic potential, clinical benefits and safety thereof, expectations regarding timing, success and data announcements of current ongoing preclinical and clinical trials, the ability to initiate new clinical programs, the risk that the results of current preclinical studies and clinical trials may not be predictive of future results in connection with current or future preclinical and clinical trials, including those for HB-200, HB-700, HB-400 and HB-500, the regulatory approval process, the timing of regulatory filings, the challenges associated with manufacturing drug products, HOOKIPA’s ability to successfully establish, protect and defend its intellectual property, HOOKIPA’s ability to achieve the expected benefits of its strategic reprioritization and other matters that could affect the sufficiency of existing cash to fund operations. HOOKIPA undertakes no obligation to update or revise any forward-looking statements. For a further description of the risks and uncertainties that could cause actual results to differ from those expressed in these forward-looking statements, as well as risks relating to the business of the Company in general, see HOOKIPA’s Annual Report on Form 10-K for the year ended December 31, 2023, as well as discussions of potential risks, uncertainties, and other important factors in HOOKIPA’s subsequent filings with the Securities and Exchange Commission, which are available on the SEC’s website at https://sec.gov and HOOKIPA’s website at www.hookipapharma.com . All information in this press release is as of the date of the release, and HOOKIPA undertakes no duty to update this information unless required by law.
Availability of Other Information About HOOKIPA Investors and others should note that we announce material financial information to our investors using our investor relations website, www.ir.hookipapharma.com , SEC filings, press releases, public conference calls and webcasts. We use these channels, as well as social media, to communicate with our investors and the public about our company, our services and other issues. It is possible that the information we post on social media could be deemed to be material information. Therefore, we encourage investors, the media, and others interested in our company to review the information we post on the social media channels listed on our investor relations website.

Contact Data
- Open access
- Published: 25 May 2024
Spatial transcriptomic brain imaging reveals the effects of immunomodulation therapy on specific regional brain cells in a mouse dementia model
- Eun Ji Lee 1 , 2 ,
- Minseok Suh 1 , 3 , 4 ,
- Hongyoon Choi 1 , 3 ,
- Yoori Choi 1 , 5 ,
- Do Won Hwang 6 ,
- Sungwoo Bae 1 , 3 , 4 &
- Dong Soo Lee 1 , 2 , 3 , 4 , 7
BMC Genomics volume 25 , Article number: 516 ( 2024 ) Cite this article
Metrics details
Increasing evidence of brain-immune crosstalk raises expectations for the efficacy of novel immunotherapies in Alzheimer’s disease (AD), but the lack of methods to examine brain tissues makes it difficult to evaluate therapeutics. Here, we investigated the changes in spatial transcriptomic signatures and brain cell types using the 10x Genomics Visium platform in immune-modulated AD models after various treatments. To proceed with an analysis suitable for barcode-based spatial transcriptomics, we first organized a workflow for segmentation of neuroanatomical regions, establishment of appropriate gene combinations, and comprehensive review of altered brain cell signatures. Ultimately, we investigated spatial transcriptomic changes following administration of immunomodulators, NK cell supplements and an anti-CD4 antibody, which ameliorated behavior impairment, and designated brain cells and regions showing probable associations with behavior changes. We provided the customized analytic pipeline into an application named STquantool. Thus, we anticipate that our approach can help researchers interpret the real action of drug candidates by simultaneously investigating the dynamics of all transcripts for the development of novel AD therapeutics.
Peer Review reports
Introduction
Central nervous system (CNS) and central immune system (bone marrow: BM) interactions, specifically brain-immune cross-talk, can occur by a pathway from the skull BM, meninges and their lymphatics, and cerebrospinal fluid (CSF) to the brain parenchyma [ 1 , 2 , 3 , 4 , 5 , 6 , 7 , 8 , 9 , 10 , 11 , 12 , 13 , 14 ] and/or by another pathway from the choroidal plexus (CP) capillary-stroma-epithelium and CSF to brain parenchyma [ 15 , 16 , 17 , 18 , 19 ] in addition to by the classic pathway of crossing the blood‒brain barrier (BBB) [ 20 , 21 , 22 , 23 ]. In explicit neuroinflammatory diseases such as multiple sclerosis in humans or experimental autoimmune encephalomyelitis (EAE) in animal model, immunoglobulins or immune cells have been considered to enter the brain parenchyma via the BBB [ 20 ] of the brain parenchyma or via the brain-CSF barrier of the CP [ 24 , 25 ], or recently via the arachnoid barrier cell (ABC) layer of skull BM-meningeal lymphatics and CSF/perivascular spaces reaching the brain parenchyma [ 3 , 4 , 5 , 17 , 26 , 27 , 28 ].
Novel immunomodulatory therapy in Alzheimer’s disease (AD) transgenic models, such as 5xFAD mice, should be accompanied by the improvement of cognitive decline associated with aging and/or the amelioration of the transgenes’ adverse effects, such as priming brain cells or immune responses during development and aging. When we inadvertently found the effect of the anti-CD4 antibody while investigating the effect of aducanumab [ 29 ] and encountered the probable effect of allogeneic natural killer (NK) cell supplements in AD models [ 30 ], we questioned which cells or transcriptomic markers in the brain areas would be the best to predict the outcome of these novel, currently unaccounted therapeutic candidates. In AD mouse models including 5xFAD mice, the surrogate effect markers of previous findings/trials of systemic or intraventricular administration of CD8 + T cells [ 31 ], anti-CD8 [ 32 ] or anti-CD3 [ 33 ] antibodies, Treg cells [ 34 , 35 , 36 ] (or for stroke model [ 37 ] or DEREG model for traumatic brain injury model [ 38 ]), and amyloid-sensitized Th1 cells [ 39 , 40 , 41 ] were amyloid plaques/Aβ on immunohistochemistry and transcriptional signatures of major brain cells and brain parenchyma [ 32 , 33 , 35 ] or CP infiltrating cells [ 25 , 42 ]. As systemically injected cells and immunoglobulins were not examined for their location or biodistribution, direct CNS effects or systemic actions on immune systems were always the alternative to explain the probable effect of novel immunomodulatory therapies, which inevitably led to the insufficient understanding of the target cells and areas. This resulted in inconsistent findings among the reporting investigators.
Single-cell or single-nucleus RNA sequencing (scRNAseq/snRNAseq) based on tissue dissociation and preparation of a single-cell suspension followed by next-generation sequencing allows comprehensive characterization of cell types in the tissue [ 43 , 44 , 45 , 46 ]. Recently available barcode-based spatial transcriptomics (ST) using the solid-phase capture of RNA on slides, such as Visium® [ 47 , 48 , 49 , 50 ], HDST [ 51 ], slideSeqV2 [ 52 ], Seq-Scope [ 53 ], or stereo-Seq [ 54 , 55 , 56 ], adds a spatial dimension to transcriptomics and enables spatial characterization of genes and cell types through robust regional segmentation of the tissue. Regional and cell-type specific characterization of one or more sections of the mouse brain based on this reliable anatomical segmentation mostly allowed for the comparison of the basal states between groups or even the task-related active states by calcium two-photon imaging and scRNAseq of the visual cortex [ 57 , 58 ]. Given that ST allows us to obtain genome-wide spatial expression profiles, ST can be considered multiplexed molecular imaging of the brain. We can now use spatial transcriptomic brain imaging to investigate whether a probable immunomodulatory therapy yields its effect on major brain cells (and infiltrating or rare cells of the brain) in each segmented brain area after systemic administration [ 30 ]. Biodistribution studies after systemic injection can inform whether immune cells or antibodies enter and directly interact with brain cells; however, if we do not see the immediate presence of cells (usually none) and antibodies (less than 1% of the injected dose), we assumed that they would influence brain cells and pathologic processes. Transcriptomic changes owing to proper novel immunomodulatory therapy will enable us to explore the probable target cells/genes that would have caused or at least be associated with the expected behavioral effects of these therapies [ 30 ]. This is especially helpful early in the pursuit of potential new drugs so that investigators can be confident that they are moving in the right direction to modify and optimize new therapeutic candidates. Transcriptomic changes of a region or regions and a cell type or cells in a group are expected to explain the behavioral results of the mouse model. Finally, the transcriptomic changes might predate the behavioral improvements. In both cases, we expect that transcriptomic profiles at a high spatial resolution would excel for drug screening, in sensitivity and target-cell specificity [ 47 , 54 ], over histological results of immunohistochemistry. Additionally, single-spot ST yields tissue-globally searchable data that can later be reanalyzed repeatedly when the marker gene combination [ 46 , 50 , 59 , 60 , 61 , 62 ] becomes available.
To do this, we needed to advance the single-spot RNA sequencing and its analyses using a customized method to derive cell type/state-specific distribution of the Visium sections. Paying attention to the proper dissociation of cell types/states using the optimum/minimum number of genes and cell‒cell interaction (CCI) and cell‒cell communication (CCC) [ 63 ], marker gene combinations should be established with the existing public database of scRNAseq/snRNAseq [ 46 , 49 , 59 , 64 , 65 , 66 , 67 , 68 ] and our own data [ 30 ]. For both tasks, ready-to-adopt methods are available by the Creative Commons regulations in previous investigations.
Stromal and parenchymal cells of various organs, including the brain, are now known to show common and specific characteristics of cell identity and their ontological characteristics, the best known of which are microglia and perivascular macrophages or resident macrophages [ 67 , 68 ]. Immune cells of monocyte-derived and resident macrophages have distinctive transcriptomic signatures, which predict their immune roles and tissue integrity-preserving roles per characteristic signatures [ 69 ]. This was also the case for T cells, where resident memory T (T RM ) cells for intestines, effector memory T (T EM/EMRA ) cells for blood, liver, and BM, and mixed T RM/EM cells for various organs and BM yield their own characteristic transcriptomic signatures that determine their differentiation of T cells in every tissue of interest, dictating their respective functional roles [ 70 , 71 ]. Unfortunately, neither of these recent cross-tissue, resident immune cell studies [ 69 , 71 ] included the brain, which mandates our own analysis.
In this investigation, we performed segmentation of brain regions on coronal/sagittal sections per dozens of animals using readily available methods and characterized the common pathologic transcriptomic signatures of 7-month-old 5xFAD mice. Immunomodulatory drugs were administered to these mice to confirm behavioral improvement. 99m Tc-hexamethylpropyleneamineoxime (HMPAO)-labeled cell-tracking imaging [ 72 ] ruled out the immediate infiltration of NK cells in the brain. Spatial transcriptomic changes in mice were examined after anti-CD4 immunoglobulin administration and expanded NK cell supplement treatment with a dose schedule, which improved Y-maze alternation behavior impairment at this age range in 5xFAD mice. Transcriptomic changes were dissected across areas and cell types/states using publicly available methods and databases, and the analytical pipelines were organized as an application named STquantool. We found that regional/areal gene set-defined type/state-specific cells showed characteristic differences after each trial treatment in a genetic model of AD, 5xFAD mice, upon spatial transcriptomic Visium analysis. Combined brain major cells including neurons, astrocytes, microglia, and oligodendrocytes with their associated types and states and brain resident/infiltrating rare immune cells per region were explored for their distinctive transcriptomic changes among mouse groups to yield their probable association with behavior improvements.
Spatial transcriptomic characterization of gene-set-defined type/state-specific major brain cells in 7-month-old wild-type and 5xFAD mice
In total, 35 coronal and 28 sagittal brain sections from 63 mice of either wild-type or 5xFAD background were included in the analysis. A spatial barcode was given for every spot, the unit tissue domain of spatial transcriptomics, and at least 50,000 reads were obtained from each of the 4,992 spots in a capture region. The brain tissues were covered by an average of 3,000 spots across all samples. Using the count matrices computed from Space Ranger as inputs to the reciprocal principal component analysis (RPCA) based integration and clustering pipelines supplied by Seurat (Seurat 4.1; https://satijalab.org/seurat/ ), the multiple brain tissues were segmented based on their transcriptome patterns [ 73 ]. By optimizing parameters such as the resolution of spatial clusters and others, we yielded segmented spatial cluster images in every case, including those with various treatments and manipulations. The treatments and manipulations are listed in Supplementary Fig. 1 A and Supplementary Table 1 . These included anti-CD4 antibody treatment and NK cell supplement treatment groups. Others were 3-month-old 5xFAD mice, cervical lymphatic ligation, a P301L model with or without amyloid/tau-rich lysate injection, and fingolimod hydrochloride (FTY720) injection with or without lipopolysaccharide (LPS) pretreatment.
The difference between 10 ST data points from wild-type and 11 from 5xFAD animals was compared for each of 14 spatial clusters using 7 to 8 coronal and 3 sagittal sections. For the integration of slides based on RPCA, the transcriptomes of wild-type animals were used as pivots, and the spots from diseased animals were mapped to the PCA space of the wild-type reference [ 74 ]. The corrected counts derived from the integration were used to cluster the spots. Spots clusters were visualized in all individual sections and verified for their accuracy in designating the areas according to already-known anatomical correlates (Supplementary Fig. 1 B-D). Only the askew sagittal section in a few mice missed the dorsal striatum and instead supplied the septal lobe in a more median position, but the spatial clustering correctly showed the pair of dorsal/ventral striatum in some sections and septal/ventral striatum in others. The spots from each cluster and group were represented by a UMAP plot, a dimensionality reduction method for visualization, and the clusters were well separated in terms of gene expression (Supplementary Fig. 1 E, F).
Using the developed platform, STquantool, the representative genes for each cell type were determined by literature or data-driven methods and validated based on spatial gene expression patterns (Materials and Methods). Cell signature scores for each cell type were calculated by the difference between the average expression of the marker genes and that of the randomly selected control genes. Since the cell scores are derived from the curated marker genes and indirectly measure the abundance of the cell type in each spot, it can be postulated that the spatial distribution of the cell scores reflects the spatial distribution of the corresponding cell type. Regional/areal transcriptomes representing major brain cell types/states were then compared between groups by averaging the major cell scores in wild-type and 5xFAD mice using the Wilcoxon rank-sum test (Supplementary Fig. 2 ). In addition to the major cell types, scores were also calculated in 32 subtypes of neurons [ 75 ], several types of astrocytes [ 76 ], microglia, and oligodendrocytes [ 66 ]. In addition, the reactive state-specific changes and marker genes of astrocytes and microglia were compared between wild-type and 5xFAD animals in 14 brain regions.
To investigate brain regional changes during pathological progression in AD, we obtained ST data from coronal brain sections of the 5xFAD mouse model and age-matched wild-type mice at three and seven months of age. The 5xFAD AD model is known to show amyloid deposition and reactive gliosis from two months of age and synaptic loss and cognitive impairment from four to six months of age (Supplementary Fig. 3 A). Amyloid deposition was observed in the adjacent sections of the samples used for ST analysis (Supplementary Fig. 3 B). In both coronal and sagittal sections, beta-amyloid levels began to increase at three months of age, and dramatic increases were observed remarkably in the deep cortex, thalamus, hippocampus, and amygdala of seven-month-old 5xFAD mice. The major brain cells were classified into neurons, astrocytes, microglia and oligodendrocytes and their associated cell types (Supplementary Fig. 4 A). Neurons were classified according to the reports of Hodge et al. [ 75 ] of the Allen Institute with Aeverman et al.’s [ 59 ] random forest hierarchical clustering method (NSForest) to define the optimal marker gene combination for neuron subtypes, which was verified by the reports of BICCN [ 77 ] and another group’s approach [ 78 , 79 , 80 , 81 , 82 ]. Astrocytes were classified according to the types of white matter-associated and gray matter-associated astrocytes [ 76 ] once and again into region-specific astrocytes for the cortex/hippocampus (telencephalon), thalamus, and other brain regions (diencephalon) [ 49 , 83 ]. Reactive astrocytes and their marker gene combinations were determined by the suggestions of Escartin et al. [ 84 ] and other investigators (Habib et al. [ 85 ], Ioannou et al. [ 86 ], Chamling et al. [ 87 ] etc.). Oligodendrocytes and their associated cell types were designated by following an initial report by Marques et al. [ 66 ] and were verified by other investigators’ suggestions [ 86 , 87 ]. Microglia were classified according to their states but not types considering their homeostatic and reactive (microglia with neurodegeneration: MgND [ 88 ], disease-associated microglia: DAM [ 89 ], lipid-droplet associated microglia: LDAM [ 90 ], etc.) states [ 68 , 91 , 92 ], and thus, no subtypes of microglia were assigned. Instead, the aging-related effect on its own or associated with amyloid pathology was examined to show amyloid pathology excluding the confounding effect of aging [ 93 ].
Using the data by Ximerakis et al. [ 64 ] for defining cell types, the hierarchical clustering method suggested by Hodge et al. [ 75 ] and NSForest (version 2.0) by Aeverman et al. [ 59 ], neurons were typed and subtyped into 20 GABAergic neurons and 12 glutamatergic neurons. Their pattern of expression is displayed for wild-type and 5xFAD mice in Fig. 1 A. The differences between wild-type and 5xFAD mice in coronal and sagittal sections of 9 areas of interest were quantitatively analyzed (Supplementary Fig. 4 B). Subtypes of GABAergic and glutamatergic neurons showed unique patterns in wild-type and 5xFAD mice. The expression patterns of the GABAergic somatostatin (Sst) subtypes in the amygdala differed between wild-type and 5xFAD mice (Fig. 1 B and Supplementary Fig. 4 C). Increased expression in the amygdala was prominent in the 5xFAD mice compared to the wild-type mice. Notably, the individual genes ( Sst, Nr2f2, Tac1 , and Moxd1 ) tended to be expressed at higher levels in the amygdala of 5xFAD mice among the gene combinations (Supplementary Fig. 5 ). Furthermore, increased expression of Sst was identified at the protein level in the amygdala and striatum regions but not in the deep cortex, thalamus, or hippocampal regions of seven-month-old 5xFAD mice (Supplementary Fig. 6 ). The most pronounced increase was found in the amygdala, with only a slight change in the striatum, and the results were consistent with those at the transcript level. Thus, after the accumulation of amyloid plaques in the 5xFAD mice, a marked increase in specific subclasses of inhibitory neuron-associated genes in the amygdala was remarkably identified.

Brain region-specific expression patterns of the signatures of neuronal subclasses and spatial changes in neurons in 5xFAD mice compared to wild-type mice. ( A ) Spatial patterns of diverse neuronal subclass signatures. The left side is a representative slide of a wild-type mouse, and the right side is that of a 5xFAD mouse. Each cell type showed distinct region-specific expression. First, mature neurons were subdivided into GABAergic and glutamatergic neurons, and then the cells were further divided into subclasses to show the regional distribution of subclasses of inhibitory and excitatory neurons. ( B ) Spatial pattern of the neuronal signatures (mature neurons, Sst1, Sst2, Sst3, Sst4, and Sst5; left). Representative images of each group were selected among 10 spatial transcriptome datasets of wild-type mice and 11 of 5xFAD mice. Spatial patterns of the Sst-subclass of inhibitory neuronal signatures of the 5xFAD mice were the most remarkably different compared to those of wild-type mice. The spatial distribution of Sst subclass neurons was similar between wild-type and 5xFAD mice, and the expression of Sst subclasses was higher in 5xFAD exclusively in the amygdala. The boxplot revealed the average module score of Sst-subclass inhibitory neurons, and expression tended to be higher in the amygdala in 5xFAD mice, especially for the Sst4-subclass. Each dot represents a mouse in each group. (mNeur: mature neurons; Sst: somatostatin; WT: wild type; TG: 5xFAD mice; GABAergicCGE: caudal germinal eminence; GABAergicMGE: medial germinal eminence; GlutamateNPCTL6b: near projection, corticothalamic, and layer 6b; GlutamateL5PT: layer 5 and pyramidal tract)
The difference between wild-type and 5xFAD mice was differential according to the definition (by gene combination to define reactivity) of reactive astrocytes and reactive microglia in their density and distribution (Fig. 2 and Supplementary Fig. 7 ). Reactive astrocytes and reactive microglia shared gene signatures and were supposed to collaborate to do the job of waste disposal in situ and out of the brain while promoting the interstitial fluid space (ISF) to perivascular/CSF space to meningeal lymphatics. Astrocytes were classified into deep or upper cortical layer-specific and telencephalon- or diencephalon-origin according to Bayraktar et al. [ 83 ] and Kleshchevnikov et al. [ 49 ]. This classification did not reveal a difference between wild-type and 5xFAD mice (Supplementary Fig. 7 A). However, another two types of astrocytes, white matter-associated and gray matter-associated, according to Werkman et al. [ 76 ], yielded differences in density and distribution between wild-type and 5xFAD mice. The cell score of white matter-associated astrocytes was significantly increased in the white matter and other gray matter regions in the 5xFAD mice, but no differences were observed in the gray matter-associated astrocyte signatures (Fig. 2 A). In addition, reactive astrocytes defined by various ways [ 84 , 85 , 86 , 87 , 94 ] that showed an increase in density in white matter and neighboring gray matter areas (cortex and thalamus) in 5xFAD mice. Their distribution of reactive states was characterized to be diffuse but was prominent around the white matter in coronal and sagittal sections from 7-month-old 5xFAD mice compared with that of wild type mice. Aging astrocytes showed significant but small differences between wild-type and 5xFAD mice in the white matter, deep cortex, thalamus, and striatum (Fig. 2 A). Further analysis with individual transcriptomes used as markers for each state-specific astrocyte revealed the following findings. The expression of individual transcriptomes defining white matter-associated and reactive astrocytes showed similar patterns between wild-type and 5xFAD mice, but the dominant individual transcriptomes were different (Supplementary Fig. 8 A, B). In the gene combination of white matter-associated astrocytes, Lyz2, C1qa, Ctss, C1qb , and C1qc were the top five genes with significant differences. In reactive astrocytes, Gfap, Serpina3n, Vim , and C1qb showed dramatic increases in 5xFAD mice compared to wild-type mice.

Spatial changes in the distribution of the region- or state-specific signatures of microglia and astrocytes in 5xFAD mice compared to wild-type mice. ( A ) Spatial pattern of the region-specific signatures (white matter-associated and gray matter-associated astrocytes) and the state-specific signatures (reactive astrocytes and aging astrocytes; left). Representative images of each group were selected among 10 spatial transcriptome datasets from wild-type mice and 11 from 5xFAD mice. Boxplot showing average module scores (right). Each dot represents a mouse in each group. The average module score of white matter-associated astrocytes was significantly increased in the white matter and other gray matter regions in 5xFAD mice compared to wild-type mice, but no differences were observed in gray matter-associated astrocyte signatures. Moreover, the average module score of reactive astrocytes showed a similar expression pattern to that of white matter-associated astrocytes, while significant but smaller differences were observed in white matter and several areas in the aging astrocyte signatures. ( B ) Spatial pattern of the state-specific signatures (plaque-associated, aging-associated, homeostatic, reactive, and panmicroglia). The average module score of plaque-associated microglia showed a significant increase in 5xFAD mice compared to wild-type mice, whereas aging-associated microglia showed no difference. Interestingly, both homeostatic and reactive microglia signatures showed a dramatic increase in the average module score in 5xFAD mice. Microglia in general (representing all the state-specific and nonspecific signatures) showed increased expression in all the regions without showing any regional distinctiveness. Bonferroni-adj. *p value < 0.05, **p value < 0.01, ****p value < 0.0001. (WM: white matter; GM: gray matter; WT: wild type; TG: 5xFAD mice)
Microglia, classified into homeostatic and reactive states [ 89 ] and aging-related and plaque-related states [ 93 ], showed increased density in wide areas for homeostatic state microglia and reactive microglia (disease-associated microglia, according to Keren-Shaul et al. [ 89 ]) and plaque-related (aging-nonrelated but plaque-related) reactive microglia. Interestingly, both homeostatic and reactive microglia showed a dramatic increase throughout brain regions in 5xFAD mice compared to wild-type mice (Fig. 2 B). Plaque-associated microglia also showed a significant increase in 5xFAD mice, but aging-associated microglia showed no difference (Fig. 2 B). Plaque-associated and reactive microglia shared a similar set of genes (Supplementary Fig. 8 C, D). In particular, Cst7, Spp1, Ccl6 , and Axl showed remarkable increases in 5xFAD mice compared to wild-type mice for both microglial signatures. For homeostatic microglial signatures, other genes, such as Hexb, Cst3, Cx3cr1, Tmem119 , and P2ry12 , showed dramatic increases in 5xFAD mice. Of note, microglial signatures did not show differences by brain region.
Oligodendrocytes and their lineage cells classified by Marques et al. [ 66 ], which comprise mature oligodendrocytes, myelin-forming oligodendrocytes, newly formed oligodendrocytes, committed oligodendrocyte precursors (COP) and oligodendrocyte precursor cells (OPC), showed distinct distribution along the areas, mainly identified in the white matter and faintly in the thalamus and lateral hypothalamus (Supplementary Fig. 7 B). A significant difference in newly formed oligodendrocytes in the deep cortex and thalamus was observed between wild-type and 5xFAD mice, but the expression was very low, and the difference was also small. The classification according to Chamling et al. [ 87 ], consisting of oligodendrocytes, OPCs and cycling progenitors, also showed similar characteristic distributions. The oligodendrocyte signatures showed relatively little difference between wild-type and 5xFAD mice.
Astrocytes and microglia, specifically white matter-associated astrocytes, reactive astrocytes, plaque-associated microglia, and homeostatic and reactive microglia, tended to increase exclusively in the white matter in 3-month-old 5xFAD mice compared to age-matched wild-type mice. This meant that the changes with the signatures started at an earlier age and occurred around white matter, reflecting a similar result to our previous report [ 95 ] (Supplementary Fig. 9 ). The most interesting finding was that homeostatic microglia also revealed increased expression in most gray matter regions at the later stage of amyloid pathology, similar to the expression pattern of reactive microglia. Increased expression of Tmem119 (a marker for homeostatic microglia) and Cst7 (a marker for reactive microglia) in the gray matter regions, especially in the deep cortex, thalamus, hippocampus, and amygdala, was validated at the protein level in 7-month-old 5xFAD mice compared to 3-month-old 5xFAD mice (Supplementary Fig. 10 A). In addition, increased expression of GFAP, S100beta, and Ctss (markers for reactive astrocytes) was confirmed at the protein level in the deep cortex, thalamus, amygdala, and white matter regions (Supplementary Fig. 10 B). Thus, the results of verifying the protein expression level were consistent with the ST analysis results.
Finally, DEGs were explored between the groups using the MAST model [ 96 ] to find regional differences at the gene level. Of note, we considered gene abundance in addition to the log fold change of mean expression in the spots corresponding to the two groups to classify DEGs in each brain region. Based on the properties of barcode-based spatial transcriptomics, adding abundance information for the corresponding gene within one barcode can increase confidence in identifying DEGs. The spatial expression of individual DEGs in 5xFAD mice compared to wild-type mice was visually assessed by STquantool (Supplementary Fig. 11 and Supplementary Table 2 ). Venn diagrams of the significantly different transcripts per region were drawn, and the GO terms associated with the genes were visualized as dot plots to examine the differences between wild-type and 5xFAD mice. The reliability of the applied DE analysis was validated by quantitative reverse-transcription PCR (qRT‒PCR analysis) in the thalamus and hippocampus (Supplementary Fig. 12 ). Among the DEGs from the thalamus, Hexb, Lyz2, Cst7, H2-K1, Ctss , and Gfap were increased in the thalamus of 5xFAD mice compared to wild-type mice. Additionally, the detected DEGs in the hippocampus, Scg5, C1qb, Ctss, Hexb, Cst3, S100a6, Cst7, Gfap , and Lyz2 , were also significantly increased. Thus, we demonstrated that our transcriptomic approach faithfully captured changes in DE analysis. In 5xFAD mice, both the white matter and gray matter regions showed significant increases in gliogenesis- and glial cell activation-related genes. For downregulated genes-associated pathways, none were detected in the white matter, but ATP biosynthetic process and purine nucleoside triphosphate biosynthetic process were significantly decreased in deep cortex of 5xFAD mice compared to wild type. The DEG-related upregulated and downregulated pathways in other regions are listed in Supplementary Table 3 .
Spatial transcriptomic characterization of rare brain-resident or infiltrating cells in 7-month-old wild-type and 5xFAD mice
Spatial transcriptomic characterization of rare brain cells poses problems of finding the proper unique set of gene combinations for determining these rare cells residing among the confounding major cells. Unlike major cells, the distribution of which is already known, rare cells are low in number and do not have any presumed distribution. Information on the propensity (rarity) of these cells is either derived from scRNAseq studies using dissociated samples from various areas of the brain, even collected from a number of animals, or from the zoomed-in small areas observed by histochemistry. Abundance studies of rare immune cells in the brain reported that the abundance of T cells was 4/mm 3 , that of neurons was 90,000/mm 3 and that of microglia was 6,500/mm 3 [ 97 , 98 , 99 ]. Other cells such as B cells, monocytes, infiltrating macrophages, dendritic cells (DCs) either conventional or plasmacytoid, or neutrophils were counted and reported for the brain tissue as a whole because all these studies were from scRNAseq analysis using suspended cells from dissociated brain tissue.
In contrast to the previous studies that disregarded the heterogeneous distribution of rare immune cells in the brain, solid-phase spot RNA sequencing enabled genome-wide quantification and localization of transcripts, as first documented by Ståhl et al. [ 47 ]. In this method and in the now available Visium, a spot has its own count (log1p of the count ratio), which was measured by in situ capture of transcripts in the tissue. However, a spot is composed of a mixture of multiple cells, and it can be difficult to distinguish the transcripts of the rare immune cells from those of the major cell types. In line with this, the problem is to find an appropriate gene (transcriptome) combination to sort out only the specific marker transcriptomes that can discern rare cells from others. Selecting the possible key gene sets defining rare cells with the highest specificity is influenced by the choice of the input data, which are composed of participating cells [ 50 ]. For example, T or B lymphoid cells, quite unique with their high propensity for ribosomal protein genes such as Rpl or Rps , are characterized by any cell-type annotation method to yield candidate marker gene combinations. However, other major brain cells are also equipped with these protein-producing genes expressed in sufficient amounts to appear to be rare brain cells, confounding the presence/density of rare lymphoid cells in any area of the brain. Additionally, since rare immune cells are commonly investigated by combining cell sorting strategies with scRNAseq, the rare cell markers acquired from the subpopulation single-cell dataset may overlap with the major cell type markers. This caused serious overestimation, which was disclosed immediately upon visual assessment. This was also the case despite the use of the recent data available by Schafflick et al. [ 68 ] and NSForest by Aeverman et al. [ 59 ]. We adopted visual curation to exclude the frankly absurd transcriptomes as marker gene candidates and finally sorted out the rare cells with optimal marker gene combinations to compare wild-type and 5xFAD mice (Fig. 3 and Supplementary Fig. 13 ).

Spatial changes in the distribution of myeloid and lymphoid cell signatures in 5xFAD mice compared to wild-type mice. ( A ) Spatial pattern of the signatures of myeloid, B cell, and T-cell and ILC compartments according to the marker gene combination reported by Dominguez Conde et al. [ 71 ] (left) and the boxplot showing the average module scores (right). Each dot represents a mouse in each group. The average module score of the myeloid compartment showed a significant increase in the white matter and gray matter regions adjacent to the white matter, including the thalamus, deep cortex, and striatum. B-cell compartment signatures showed an increase in the deep cortex and white matter. In contrast, T-cell/ILC compartment signatures were low without differences between wild-type and 5xFAD mice. ( B ) Spatial pattern of the subtype signatures of myeloid cells, including CAM, macrophage, monocyte, plasmacytoid DC, and granulocyte according to marker gene combination from Schafflick et al. [ 68 ]. Notably, CAM and macrophage signatures showed the most pronounced increase in 5xFAD mice compared to wild-type mice in most of the regions. Monocytes and plasmacytoid DCs were increased in the deep cortex and white matter, and plasmacytoid DCs were further increased in the thalamus, pyriform area and striatum. ( C ) Spatial pattern of the subtype signatures of lymphoid cells, including NK and T cells, according to marker gene combination from Xiemrakis et al. [ 64 ]. In the case of the NK cell signature, a significant increase was observed in the deep cortex of 5xFAD mice, which is associated with an increase in CD56dim NK cells. Among T-cell signatures, tissue-resident memory T-cell signatures were higher in 5xFAD mice in the deep cortex, white matter, thalamus, pyriform area and striatum. The CD4 signature was explicit in the striatum in both wild-type and 5xFAD mice, but the expression was too low to show a quantitative difference between wild-type and 5xFAD mice. Bonferroni-adj. *p value < 0.05, ***p value < 0.001, ****p value < 0.0001. (WT: wild type; TG: 5xFAD mice; ILC: innate lymphoid cells; CAM: CNS-associated macrophage; DC: dendritic cells; NK: natural killer)
Immune resident cells were classified in three ways (1) using novel data by Eraslan et al. [ 69 ] (Supplementary Fig. 13 A) for tissue-specific monocyte-derived macrophages and data by Dominguez Conde et al. [ 71 ] (Fig. 3 A) for tissue-resident T cells, (2) using the data by Schafflick et al. [ 68 ] (Fig. 3 B) and markers refined using NSForest 2.0 and (3) using the data by Xiemrakis et al. [ 64 ] and refined using NSForest 2.0 by Aeverman et al. [ 59 ] (Fig. 3 C). The first two reports [ 69 , 71 ] were derived by using various tissues excluding the brain, while the other two reports [ 64 , 68 ] were derived by using brain tissues.
Defining marker gene combinations was more intricate for these rare immune resident/infiltrating cells, as they were defined by surface markers in the report of Eraslan et al. [ 69 ], in organs/tissues other than the brain, or by transcriptome signatures suited for each study. Although the data were from body tissues, not the brain, in the first approaches, as the tissue stromal cells are included in DEG analysis and assuming stromal cells might be more similar between tissues including brain, transcriptomes of major parenchymal/stromal cells coexpressing with rare immune cells were to be correctly excluded. We chose NSForest to help exclude confounding stromal tissues. Monocyte-derived macrophages include two types, according to Eraslan et al. [ 69 ]: one for immune function (MHCII+) and the other (LYVE1+) for vascular integrity and repulsion of infiltrating immune cells. For immune function, specifically for the brain, conventional DCs with MHCII + cells were found to be effective for antigen presentation by adaptive immune cells (T cells and B cells), although border-associated macrophages or microglia were not [ 100 ]. We asked whether this surface marker-defined characterization can be translated to mouse (5xFAD) amyloidosis using the signature of MHCII+-related immune-functioning macrophages and LYVE1+-related integrity-charged macrophages described by Eraslan et al. [ 69 ] (Supplementary Fig. 13 A). Integrity-charged macrophage signature scores were not different among the groups of 7-month-old wild-type and 5xFAD mice in all regions of brain. Spot signatures of immune macrophages were higher in 5xFAD mice than in wild-type mice in the white matter.
Signature gene combinations used in the cross-tissue immune cell analysis by Dominguez Conde et al. [ 71 ] revealed no difference between wild type and 5xFAD mice for T cells and innate lymphoid cell T/ILCs. However, significant differences were found among these mice for myeloid compartment cells in the white matter and the gray matter regions adjacent to the white matter, including the thalamus, deep cortex, and striatum and for B cells in the deep cortex and white matter (Fig. 3 A).
This result came from the following stepped analysis including the curation procedure. At the first step of curation, individual transcriptomes belonging to the three compartments described by Dominguez Conde et al. [ 71 ] were examined visually for their distribution/intensity, and several transcriptomes that were already reported in the literature as signatures for major brain cells and their reactive states were removed, which excluded the background effects of abundant brain cells, eventually yielding marker gene combinations for the three compartments and their cell types. Having removed (1) Cx3cr1 and Tyrobp (microglia) from T/ILCs, (2) Ighm (Scheurer et al. [ 101 ] for neurons) and C1qa (microglia) from the B-cell compartment, and (3) Trem2 (microglia), Clu (astrocytes), Selenop (microglia, astrocytes, oligodendrocytes), Igf1 (reactive microglia and reactive astrocytes), C1qa and Cx3cr1 from the myeloid compartment, the scores of T/ILCs were still not different between wild-type and 5xFAD mice, and the scores of B-cell or myeloid compartments revealed a slight but significant increase in 5xFAD mice compared with wild-type mice. Individual variations within 5xFAD mice could also be recognized upon visual assessment. For individual genes for T/ILCs, localization was prominent for Cd4 (striatum) and showed little difference regardless of abundance ( Slc4a4, Spry2, Ncam1 , and Pcdh 9 are abundant), and no difference was observed except for Pdcd1 (smaller cell fraction in various T cells including Trm/em_CD8 according to Dominguez Conde et al. [ 71 ]), which was slightly increased in 5xFAD mice. For the B-cell compartment, the difference between mouse groups, if any, was presumed to be due to Itgax and Fcrls , both of which were related to aging-associated B cells, and Fcrls was related to memory B cells and plasma cells/plasmablasts. For the myeloid compartment, the difference in scores among mouse groups was contributed by Tyrobp, Lyz2, Fcer1g, C1qc , and Apoe , all of which are related to various types of tissue-specific macrophages and classical/nonclassical monocytes (Supplementary Fig. 14 ).
The second one by Schafflick et al.’s data [ 68 ] was tested for either the marker gene combination recommended by Schafflick et al. according to their supplementary table (log fold change: LFC > 0.5) for 12 border cell leukocytes (including microglia) or the marker gene combination curated by NSForest upon inputting their data. Schafflick’s own data yielded obviously too high intensity for CD4 and CD8 T cells among 12 border-associated leukocytes, such as B1, B2, CD4 T, CD8 T and NK cells, microglia, CNS-associated macrophages (CAM), macrophages, monocytes, myeloid DC (mDC), plasmacytoid DC (pDC) and granulocytes. When we surveyed the constituents of the tentative marker transcriptomes for these inappropriate signatures, ribosomal genes (many isoforms of Rpl and Rps ) were presented as false positive markers of CD4 T and CD8 T cells. This misclassification of marker genes is assumed to be caused originally by the fact that the parenchymal and border leukocytes were included after their selection for CD45 positivity, meaning that they could not exclude the differential expression of these cells from the major brain cells, including stromal cells. Individual transcriptomes per spot were easily observed to disclose whether we chose the highest LFC with adjusted p values for determining marker genes, Ighm for b1 cells (also found in the cortex not related to B cells) [ 101 ], H3f3b (histone protein also nonspecific for the brain) for b1 cells, Stmn1 for b2 cells (rather brain-wide expression), Dut (enzyme for nucleotide and ubiquitous, including brain cells) for B cells and many similar examples (Supplementary Fig. 13 B). Although DEG analysis depends upon the input data composition, we tried NSForest on Schafflick’s data and obtained a better marker gene combination. This Schafflick/NSForest analysis yielded improved intensity matching considering the prevalence of cell populations in the brain parenchyma except for b2 cells (still too dense due to Tuba1b (tubulin related)) and CD4 T cells (depending heavily upon one transcriptome Trbc2 (T-cell receptor beta constant 2 but also expressed in the cortex)). The other 10 cell signatures appeared to represent the cell intensity/distribution correctly; however, they also included nonspecific and dense Apoe for CAM, dense Cst3 for macrophages, Mal (Myelin And Lymphocyte Protein, implying its localization both in lymphocytes and myelin of neurons) for monocytes, and Tyrobp (in association with Trem2 , a well-known marker for microglia) for both monocytes and pDCs. Upon the application of NSForest, 6 to 10 marker genes were obtained, and zero to three genes were adjusted (kept or removed, meaning curated by operators’ consensus). The application of curated gene combination to our ST samples revealed that CAM and macrophages showed the most pronounced increase in 5xFAD mice compared to wild-type mice globally throughout the brain regions (Fig. 3 B). Additionally, pDCs showed increases in the white matter and some gray matter regions. However, the transcriptome density of DCs was considered inappropriate, as it yielded much higher intensity along the entire brain, considering that DCs occupy only 0.14% of myeloid/lymphoid cells of the brain and border, including microglia (0.8% among myeloid/lymphoid cells excluding microglia) [ 68 ].
Among lymphoid cell signatures, an increase in tissue-resident memory T cells was inferred in 5xFAD mice compared to wild-type mice (Fig. 3 C and Supplementary Fig. 14 ). However, it is necessary to consider the technical limitations of spot-based transcriptomic analysis for evaluating rare brain cell signatures. It is still unclear which genes specifically define rare cells, while gene combinations may overlap with major brain cells on ST brain imaging.
Using a single gene as a marker would be better and more convenient for designating rare cell types. It was possible to designate infiltrating macrophages derived from circulating monocytes originating from BM (Supplementary Fig. 15 ). The CD11c surface marker and its gene Itgax were used as markers for these cells. Resident T cells were suggested to be CD73 positive, and its gene Nt5e was identified by Fang et al. [ 102 ]. CD56 bright and its gene Ncam1 are considered to be circulating and immature NK cell markers but are also highly expressed in neurons [ 103 , 104 ]. Perivascular macrophages cause a great problem in distinguishing them from microglia, and Lyve1 is the discriminator of pvMϕs and microglia ( Sall1 ) [ 105 ]. Similarly, for brain major cells, Trem2 and Tyrobp were suggested to be conjoint markers for microglia, and Cspg and Olig2 were expected to represent OPCs, not any other cell types. Homeostatic microglia could have been defined by Sall1 ; however, a transgenic mouse study [ 105 ] found that Hexb was the better marker for authentic microglia than Sall1 . The importance of Aif1 (IBA1) as an activated microglial marker and of Gfap as an activated astrocyte marker was disclosed to be nonspecific or at least subtype specific, respectively. Once a marker was well defined for designating rare cell types well discriminated from major brain cells, including microglia and perivascular space (pv) macrophages (and submeningeal macrophages), then that marker in a spot could disclose the fact that the gene signature of that spot might be from the rare cell of interest, but it does not mean that the signature was not from the rare cells if no signal was observed. Genes widely expressed over all cell types but with specific isoforms could be used to define the cell types, and Prdx (for peroxiredoxin) was one of the examples ( Prdx6 and Prdx2 for astrocytes, Prdx4 for microglia and Prdx1 for oligodendrocytes) (Supplementary Fig. 16 ).
To validate the results obtained from the cell signature score based method of measuring cell type abundance, we performed the cell type deconvolution analysis and compared the results between the two methods. The cell type deconvolution method captures the gene expression patterns of cell types from the single-cell reference dataset and predicts the cell type composition in the ST spot, which is a mixture of multiple cells. We performed the analysis mainly for microglia and infiltrating immune cells, which showed significant changes between 5xFAD mice (TG) and wild-type mice (WT) in the signature score-based method. For microglia, the proportions of both homeostatic and reactive microglia were globally increased in the gray and white matter regions of TG mice, which was consistent with the results obtained from the cell signature scores (Supplementary Fig. 17 ). Minor immune cells, including myeloid cells such as macrophages, monocytes, and dendritic cells, were also upregulated in multiple gray and white matter regions of TG, and the results were similar to those obtained using cell signature scores (Supplementary Fig. 18 A, B). However, for lymphoid cell types such as innate lymphoid cells, natural killer cells and T cells, which are rare, the patterns of change between the two methods were inconsistent for a few gray matter regions, while the biological effect may be small due to very low cell abundance (Supplementary Fig. 18 C, D). Overall, the results suggest that cell signature scores derived from curated markers are an accurate and reliable measure of cell abundance for the relatively common cell types, while rare cell types require special attention in interpretation.
Improvement of behaviors with much variation by immunomodulatory therapy of anti-CD4 antibody and NK supplements in the 5xFAD AD mouse model
During a preliminary behavioral study to prove the effect of aducanumab, pretreatment with anti-CD4 antibody caused a larger degree of changes in alternation scores in the control animals (meaning higher improvement in the group of animals treated with anti-CD4 antibody) [ 29 , 30 ]. Three batches of several animals with anti-CD4 antibody treatment reproduced the previous groupwise behavioral improvement with similar variation (67.7% ± 18.4%) at 7 months of age in 5xFAD model mice (Fig. 4 ). We assumed that anti-CD4 antibody treatment modulated the systemic adaptive immune system, as transgenic insertion of five types of mutated human APP/PS1 genes would have caused immune disturbance due to their presence in the mouse chromosome. The presence of human mutated genes would have resulted in brain-immune interaction dysfunction as well as plaque-prone amyloid burden in animals. Spatial transcriptomic analysis was considered to reveal the eventual response of brain cells, either major or rare resident and infiltrating immune cells, if any.

Improved behavior after intravenous administration of NK cell supplement and anti-CD4 antibody in 5xFAD mice. ( A ) Timeline of the experiments for intravenous NK cell supplements (upper) and anti-CD4 antibody (lower) administration in 6.5-month-old wild-type and 5xFAD mice. NK cells (2 × 10 6 cells/injection) were administered once a week for a total of five times, and anti-CD4 antibody (0.5 mg/injection) was administered once as a single injection. After a month, behavior analysis was performed, and brain tissue samples were obtained for spatial transcriptomic brain imaging analysis. ( B ) The behavioral function of exploring new environments was examined using the Y-maze test and expressed as alternating percentages. Each dot represents a mouse in each group. The alternation rate was decreased in 5xFAD mice compared to wild-type mice, with much variation at this age in wild-type and 5xFAD mice. The alternation percentage score of 5xFAD mice increased significantly after injection of NK cell supplements and anti-CD4 antibody treatments compared with that of 5xFAD mice without treatments. Wild-type mice also showed variation; however, their alternation scores were not different between the no treatment and either treatment group. Wilcoxon *p value < 0.05, **p value < 0.01. (aCD4: anti-CD4 antibody; WT: wild type; TG: 5xFAD mice; WT_NK: NK cell-treated wild type; WT_aCD4: anti-CD4 antibody-treated wild type; TG_NK: NK cell-treated 5xFAD; TG_aCD4: anti-CD4 antibody-treated 5xFAD)
In another preliminary study with APP/PS1 model mice using the water maze with expanded NK cell supplements derived from the spleen of wild-type BALB/c mice, anecdotal cases of behavioral improvement were observed (data not shown). Three batches of allogeneic NK cell supplements, as 5xFAD mice are on a B6 background, reproduced the behavioral improvement of alternation scores on Y-maze tests on average, however, with much variation (Fig. 4 ). Much variation in both the anti-CD4 antibody treatment study and NK supplementation study indicated that 5xFAD mice at 7 months of age were undergoing their own course of aggravation of the pathological changes of Aβ oligomer insults and amyloid plaque burden, resulting in later pathological and behavioral dysfunction at approximately 12 months of age or later. Spatial transcriptomic analysis was also considered to reveal the regional and cell-type specific changes of transcriptomes of major and rare brain cells corresponding to each individual mouse’s degree of behavioral dysfunction in the NK supplement-treated group.
Regional cell-type/state-specific transcriptome changes in 5xFAD mice compared with wild-type mice after intravenous administration of NK cell supplements
Three mice with higher alternation scores on the Y-maze test were selected for both the saline-treated and NK cell supplement-treated groups (Supplementary Fig. 19 ). Coronal/sagittal brain sections of these mice were subjected to Visium analysis. Each group was paired to the same plates so that the batch effect of the read per slide would be minimized. Using 30,000 to 50,000 counts per mouse, we retrieved the count data, which were normalized for their total count, and log1p of the ratio data were used for further analysis. Spatial clustering allowed anatomical segmentation to yield 14 regions with almost similar sizes (Supplementary Table 4 ). Cell type- and state-specific marker gene combinations were also used to analyze cell-specific and/or cell state-specific changes after NK cell supplement treatment. For the 5xFAD case with NK cell supplements, one mouse with a very high behavior score was chosen as the ‘behaviorally best’ representative of the group, and another mouse with a very low score was chosen as the ‘behaviorally worst already at 7 months of age’ representative. This essentially allowed us to examine the transcriptomic changes according to the behavioral impairment of the 5xFAD mice at the age of 7 months. NK cell supplements contributed at least to the widening of the distribution of scores of behavioral impairments at this middle age.
GABAergic Sst subtype neurons showed a significant decrease after NK supplementation in the amygdala, which showed an abnormally increased signature in 5xFAD ( n = 11) compared with wild-type mice ( n = 10) (Fig. 5 A). Among the Sst neuronal signatures, Sst, Tac1 , and Nr2f2 showed dramatic decreases in the amygdala after NK cell supplement administration in 5xFAD mice, but there were no distinct differences in the other regions (Supplementary Fig. 20 A, B). The Sst-expressing neurons in the cortex are known to contribute to modulating cortical circuits, synaptic plasticity and maintaining spatial working memory [ 106 ]. Patients with AD exhibited low Sst expression in the cortex and hippocampus. However, the function of Sst-inhibitory neurons in the amygdala remains poorly understood [ 107 ]. No significant difference was found either in the mature neuron score or in any other subtype of neurons other than Sst neurons between 5xFAD mice without NK supplements and those with NK supplement treatment. Thus, normalization of excitatory and inhibitory neuronal imbalances in the amygdala may improve behavior function. Further investigation of the neurons in the amygdala region could play an important role in understanding the pathology of AD and in providing therapeutic directions. Additionally, the NK cell signature tended to increase after administration of NK cell supplements exclusively in the white matter region of 5xFAD mouse brains (Supplementary Fig. 21 A). The change in the module score level was not observed with the anti-CD4 antibody treatment, which was in contrast with the change after NK cell supplement treatment (Supplementary Fig. 21 B). The signatures of astrocytes, microglia, and oligodendrocytes did not show any difference. Additionally, no difference in rare brain cells, either resident or infiltrating, was observed (Supplementary Fig. 22 ). The biodistribution of 99m Tc-HMPAO-labeled NK cells was examined using SPECT/CT to determine how systemically injected NK cells caused changes in the brain (Supplementary Fig. 23 ). Within 1 h after injection, the labeled NK cells were mainly taken up by the liver, and this radioactivity decreased gradually by 16 h. Of note, no definite brain uptake of the labeled NK cells was observed with the resolution of SPECT/CT images. Thus, NK cells may have caused changes in brain cells at the transcriptional level indirectly via cytokines or other secretory factors released by NK cells and/or inherent peripheral immune cells influenced by supplemented NK cells.

Brain region-specific transcriptome changes in cell signatures after NK cell supplementation and anti-CD4 antibody treatment in 5xFAD mice. ( A ) Spatial pattern of the signatures of somatostatin (Sst)-inhibitory neuronal signatures (Sst1, Sst2, Sst3, Sst4, and Sst5; left) and the boxplot showing the average module scores in the amygdala (right). Each dot represents a mouse in each group. The average module scores of Sst neuronal subclasses tended to decrease specifically in the amygdala after administration of NK cell supplement in 5xFAD mice. Interestingly, the Sst neuronal signatures, which were increased in expression in the amygdala of 5xFAD mice, were decreased to the expression level in wild-type mice by NK cell supplements. In contrast, the module score was not different between the no treatment and anti-CD4 antibody treatment groups, while there were differences between the no treatment and NK cell supplement groups. ( B ) Spatial pattern of the signatures of state-specific glial cells (aging astrocytes and reactive microglia; left) and ( C ) immune cells (monocytes and plasmacytoid DCs) and the boxplot showing the average module scores in the white matter (right). The expression of state-specific subtypes of glial cell and immune cell signatures, which showed a significant increase in 5xFAD mice compared to wild-type mice, tended to slightly decrease in the white matter after anti-CD4 antibody treatment. Considering that NK cell supplementation showed no appreciable differences in glial cell and immune cell signatures, anti-CD4 treatment effects on these cell subtypes of state-specifics looked real. Expression, however, did not decrease to the level observed in wild-type mice. In summary, NK cell supplementation and anti-CD4 antibody treatment affected different state/type-cell signatures and brain regions, respectively. (aCD4: anti-CD4 antibody; WT: wild type; TG: 5xFAD mice; WT_NK: NK cell-treated wild type; WT_aCD4: anti-CD4 antibody-treated wild type; TG_NK: NK cell-treated 5xFAD; TG_aCD4: anti-CD4 antibody-treated 5xFAD; Sst: somatostatin; DC: dendritic cells; NK: natural killer)
Regional cell-type/state-specific transcriptome changes in 5xFAD model mice compared with wild-type mice after intravenous anti-CD4 antibody treatment
Three mice in the anti-CD4 antibody treatment group were selected, and their coronal brain sections were plated on Visium slides. The frame of sample distribution on the quadrants of each Visium slide was the same as above for NK cell supplement treatment. Further analysis of transcriptomes per spot and spatial clustering and designation of transcriptomes to the approximately 3,000 spots were also the same.
Among major brain cells, state-specific glial cells, such as aging astrocytes and reactive microglia, which showed a significant increase in 5xFAD mice compared to wild-type mice, showed a slight decrease exclusively in the white matter after administration of anti-CD4 antibody in 5xFAD mice (Fig. 5 B). In the gene combination of aging astrocytes, the expression of Lgmn, Gsn, Mt1, Fcrls , and Hexb was noticeably decreased in the white matter after anti-CD4 antibody administration in 5xFAD mice, and expression decreased slightly in the other regions (Supplementary Fig. 20 C, D). In the reactive microglial signature, Cst7, Spp1 , and Cd9 showed a decreased pattern throughout the region, while other genes, such as Axl , Csf1 , and Ccl6 showed decreased patterns mainly in the white matter (Supplementary Fig. 20 C, D). Interestingly, the CD4 T-cell signature tended to decrease slightly in the deep and upper cortex (striatum) after anti-CD4 antibody treatment (Supplementary Fig. 21 B). However, mature neuronal signatures showed typical and very similar patterns of distribution between 9 major brain regions and within each region irrespective of whether the sample was from wild type, 5xFAD, wild type with anti-CD4 antibody treatment or 5xFAD with anti-CD4 antibody treatment mice (Supplementary Fig. 22 A). No significant differences in other types of glial cells, including white matter-associated astrocytes, reactive astrocytes, plaque-associated microglia, homeostatic microglia, and oligodendrocytes, were found, meaning that all 10 wild-type mice and all 11 5xFAD mice could not be differentiated between no treatment and anti-CD4 antibody treatment (Supplementary Fig. 22 B, C). The difference between wild-type and 5xFAD mice was sustained but did not reveal any dramatic effect of anti-CD4 antibody treatment.
Rare brain cells, resident or infiltrating, were distinguished between the no treatment group and the anti-CD4 antibody treatment group. The expression levels of monocytes and pDCs, which showed a significant increase in 5xFAD mice compared to wild-type mice, tended to slightly decrease only in the white matter after anti-CD4 antibody treatment (Fig. 5 C). In both monocyte and pDC signatures, the expression of Tyrobp was dramatically reduced throughout the brain, whereas S100a4, S100a10 , and Mal in the monocyte signature showed decreased expression patterns exclusively in the white matter after anti-CD4 antibody treatment. A reduction in the expression of individual genes by the anti-CD4 antibody was mainly identified in the white matter region (Supplementary Fig. 20 C, D).
While NK cell supplementation showed no appreciable differences in immune cell signatures, the fact that anti-CD4 antibody treatment showed effects on subtypes of immune cells is noteworthy. However, the expression level was not decreased to that observed in wild-type mice.
Methods to scrutinize spatial cell-type and cell-state specific changes upon the platform of setting norms and characterization of abnormality of a test mouse
As spatial distribution is critical for characterization of a new mouse specimen for their status of normalcy, pathology, and response to therapy, the specimen can be on every section, but on a limited number of coronal sections (per monkey samples in a report by Chen et al. [ 55 ]) or sagittal sections. To acquire representative information regarding mouse groups, we combined multi-individual mouse sections to yield the apparently correct spatial segmentation. Each region was then prepared to present their norms for various scores for cell types, cell states and response to the tentative immunomodulatory drugs. We tried to establish methods to reveal the regional cell-type/state-specific norms and their probable changes by drug intervention. We set up norms for normal mice using wild type mouse data genotype, and then the effect of the age or the influence of drug treatments were characterized. For example, the presence of anomaly were examined for individual mice according to their disease states (5xFAD mice of certain age with diverse behavior/Aβ abnormality, P301L mice with no behavioral/pathological abnormality) and the effect of therapy (anti-CD4 antibody or NK cell treatments).
Cell types should have been annotated to the then-best knowledge of the scientific community based on the resource reports in the literature up to the date of this investigation run by trial-and-evaluation and then the choice; for example, for neurons and neuronal subtypes, Hodge et al.’s report [ 75 ] was adopted as is or after NS Forest [ 59 ] to define 20 GABAergic neurons and 12 glutamatergic neurons. Available data were downloaded from specific sites or supplementary tables of each report. Thus, for example, Scng-VIP neuron subtypes described in a more recent report by Bugeon et al. [ 57 ] were ignored but later can be reanalyzed with the current Visium data by specifying their markers along the regions segmented after integration of slides using RPCA. For astrocytes and microglia, the reactive state signature was surveyed by scrutinizing the counts of transcriptomics at each spot according to the reports by Keren-Shaul et al. [ 89 ], Friedman et al. [ 91 ], Grubman et al. [ 93 ] and others. Coexpression of the same transcriptome by astrocytes and microglia, such as Apoe, Gfap, Tspo and others, was removed from the tentative marker gene combination. The same procedure was performed for astrocytes and oligodendrocytes or microglia and oligodendrocytes. Oligodendrocytes and their lineage cells did not have a ‘reactive’ transcriptome signature. Signature transcriptomes between reactive and homeostatic microglia (and astrocytes) were also surveyed for their conjoint expression between both states. Homeostatic transcriptomes were designated to exclude the signature of reactive transcriptomes and vice versa, referring to literature reports by Prinz et al. [ 108 , 109 , 110 , 111 , 112 , 113 , 114 ], and Kim et al. [ 105 ], so that Sall1 and Hexb were used to measure the abundance of microglia as each microglia express these genes constitutively while assuming that these transcriptomes did not increase in quantity per microglia when reactive [ 105 , 113 ]. As spatial transcriptomic imaging using Visium yielded a linear (semi)quantitative (due to log1p transformation for further processing using Seurat 4.1)) metric, no fractional presentation was adopted to disclose that homeostatic microglia were increased in quantity of signature per spot in 5xFAD mice compared with wild-type or P301L mice.
Quantification was performed for 9 regions (hypothalamus, thalamus, deep cortex, white matter, upper cortex, hippocampus, amygdala, piriform area, and striatum) for major cell types, including neuron subtypes, reactive and homeostatic glial cells, astrocyte subtypes, oligodendrocyte lineage cell subtypes and rare resident/infiltrating immune cells. For regions, for example, white matter-associated astrocytes or white matter-localized microglia were quantified and correlated with white matter-localized oligodendrocyte lineage subtypes. Dense and thus intense quantities of mature oligodendrocytes could be compared among mouse groups. In contrast, diffuse and sparsely distributed rare immune cells were found in three compartments by Dominguez Conde et al. [ 71 ], four types by Xiemerakis/NSForest [ 59 , 64 ], and 12 types by Schafflick et al. [ 68 ]. Tissue resident macrophages by Dogra et al. [ 115 ] and Eraslan et al. [ 69 ] were also quantified for intensity to yield the difference for each region among mouse groups. The Wilcoxon rank-sum test was used to determine the significance between pairs of groups (uncorrected p value or corrected by three for paired group comparisons). Beyond group comparison, an anomaly detection procedure (or confirmation of normalcy meaning no difference found on any regional, cell-type, cell-state specific signature by density per region) was performed for each mouse.
Once a region and cell type and its state were found, we performed DEG analysis to find the transcriptomes of interest in each brain region. Then, the association of discovered genes with biological pathways was examined using an overrepresentation test based on evidence-driven databases. This was to determine the significance of the found transcriptome designating their functional role in pathology (amyloid pathology or tauopathy, glial cell dysfunction, etc.), physiology (aging) and their participation in the response to tentative immunomodulatory therapy. Considering the diversity of behavior improvement after anti-CD4 antibody and NK cell supplement treatments and the unpaired nature of the Visium study, we could only detect the treatment effect (and non-effect) of the transcriptome signature of regional cell types/states upon treatment per individual. Transcriptomes of the marker gene combination that we used were all checked for their individual transcriptomes to elucidate key transcriptomes for specifying type/state characteristics or therapy effects. We also tried to assess their individual contribution to this specification to find one, two or more distinctive transcriptomes to predict their presence in each spot. This means that curation by operator in addition to the readymade Wilcoxon, logistic, or NSForest methods was used in at least two steps, first to choose a seemingly optimal combination ruling out cross-expressing, background, or confounding genes and then at last to find the succinct combination of transcriptomes for cell-type/state annotation or if any, the sole transcriptome (Supplementary Fig. 2 ). The above pipelines for dissecting cell-type- and cell-state-specific regional transcriptomic changes can be readily implemented with our in-house application STquantool, which facilitates the visualization of spatial gene expression and enables quantification across multiple transcriptomic datasets.
In this investigation, we used ST for its superiority over scRNAseq/snRNAseq to localize the specific transcriptomic signature of cell type or cell state in almost 5 thousand spots, among which 3,000 or more spots harbored either coronal or sagittal sections of brain tissue. Before going further to use this transcriptomic signature to disclose the effects of novel but unproven neuromodulatory treatments, we trimmed the method of the use of this Visium-based ST imaging to elucidate regional, cell-type/state-specific changes. The method for choosing one or two optimal transcriptomic marker combinations among so many possible combinations was adjusted to yield the best contrast between cell types/subtypes using the literature resources and our in-house method of curation. A simple and easy method to sort out the candidate transcriptomes was set up to ensure that we found the best cell type/state annotation methods for either abundant brain cells or rare immune cells. The challenge was to separate 4 or more major brain cell types and their subtypes with transcriptome combinations and to define rare immune cells for their exact propensity and distribution/location. Stahl et al.’s [ 47 ] suggestion of counting the transcriptomes per spot using the original ST Visium methods and Tirosh et al.’s [ 44 ] approach to generate cell signature scores based on the curated marker genes and comparing them between mouse groups with genuine or sham treatments worked well for this endeavor. We overcame the problem of high dependence of this endeavor on the choice of tentative marker gene combinations varying upon the diverse input data derived from the preliminary DEG studies using single-cell data of brain tissues [ 67 , 68 , 108 ] and even tissues other than brain tissues. Assessing the sophisticated use of the public database and scrutinizing the individual transcriptomes visually by the operators (neuroimaging experts) were essential. Curation by operators is heuristic at best and is surely subject to operator arbitrariness; however, this was eventually the key step to enhance the authenticity of the observation of large number of cells (2 to 10 per spot) admixed in spots and a dozen specimens from individually different but syngeneic mice. From the neuroimaging perspective, integrated single spot imaging (100 µ x 100 µ x 10 µ) containing an average of 5 cells (2 to 10) in each unit domain did not have a significant batch/individual variation effect to confound further analysis, as we observed dozens/hundreds of spots at the same time, and the batch effect was corrected during sample integration. With this visual investigation, we soon became confident that spatial transcriptomic brain spot imaging with visual assessment and its quantitative analysis using the framework of voxel (spot) imaging of mouse/human brains was suitable for the evaluation of the effect of certain drugs/treatments for disease-course modification in dementia mouse models.
The transcriptomic signature of brain cells could clearly segment every section from the mice, regardless of disease or treatment status, taking advantage of 22,000 or more transcriptomes per cell to identify the cell type/state with thousands of variable transcriptomes. Unlike functional neuroimaging such as functional magnetic resonance imaging (fMRI) or positron emission tomography (PET), which needs coregistration and segmentation considering individual variation for further analysis, the segmentation of neuroanatomical entities on Visium could be performed without any more assumptions, except that functional regional entities could be determined by transcriptomes belonging to spots and their conglomerated spots make explicit functional regions. An eccentric case of regionally remote but similar transcriptome composition was observed in that the cortical amygdala and subcortical septal lobe were categorized as the same cluster on sagittal section, but excluding this exception, all the other spatial clusters were within the expected anatomical border definition (https://connectivity.brain-map.org/3d-viewer?v=1&types=IMAGEPLANE&IMAGEPLANE=imageplanes). Thus, spatial or regional representation of characteristic changes related to pathology and treatment response could then be described and quantified. Finding marker gene combinations to define the spots as belonging to certain functional regions of interest then could be achieved by finding the optimal or best combination, which would be appropriate and succinct.
Determination of the best annotation of neurons and other major brain cells was initially dependent upon previous reports mainly derived from nonspatial scRNAseq/snRNAseq analysis [ 63 , 73 , 80 , 116 ]. In these previous studies, the spatially expected designation of cells was suggested as a success of cell clustering, raising concerns that there was no gold standard information regarding their true location; nevertheless, the cell clustering and annotation allows assignment of regional identity of brain cells based on anatomical region-specific marker genes. ST brain imaging obviated this concern. In ST imaging, however, there remain two major problematic ambiguities for spatial clustering and cell type/state identification per spot. The first one is spatially agnostic annotation by transcriptome signature, which previous researchers tried to solve by sampling regions of brain such as posterior isocortex, hippocampus (or hippocampal formation), striatum, thalamus and hypothalamus, etc., in the reports of Saunders et al. [ 65 ] or Chai et al. [ 117 ]. This problem was easily solved by ST imaging using Visium of 3 to 5 thousand spots, which allows capturing transcriptomic changes across the broad area of the brain. Now, imaging with a resolution of 100 µ x 100 µ on 2D is available, allowing easy segmentation; this differs from fMRI/PET in that the huge multiplexing capability of ST brain imaging allows almost infinite reanalyses using combinatorics. The second is cell/state identification per spot by using the transcriptomic signature of the marker gene combination determined by previous DEG studies using detached and sometimes surface-marker-sorted brain cells. When scRNAseq/snRNAseq was used for detached brain cells to determine the effect of drug/treatment on those brain cells, lack of spatial localization was the major hurdle blocking the understanding of the role of any treatment. In situ hybridization of immunohistochemistry complemented transcriptomic global/regional brain signatures to address this, but without reassuring results to explain the therapy effect. ST brain imaging solved this problem. As shown in this study, ST brain imaging is equipped with the expression profile per spot for the entirety of genes of the individual cells localized on each spot, and the data could be analyzed in an unsupervised fashion without any assumption or in appropriate cases by using a priori knowledge derived from the literature resources of scRNAseq/snRNAseq. Considering the challenges and difficulties in overcoming these problems, we streamlined the use of visual reading by expert operators called curation. The steps required for curation were kept minimal and practical, and it was performed initially to exclude nonspecific and cross-expressing transcriptomes between major cells, and finally to exclude cross-tissue, stromal cell-dependent and confounding background signatures. It would have been better to base curation on individual transcriptomic features of any types of cells for their association with disease states or drug/treatment responsiveness.
To tackle these problems, we asked how we could use individual mouse ST brain imaging data to determine the disease states, which are variable even in syngeneic animals, and the variable treatment responses affecting major and rare brain cells. Taking advantage of the automatic segmentation results for groups of individual mouse specimens, irrespective of section planes and stereotaxic coordinates, we tried to individualize the transcriptomic features of each individual specimen compared with the norms we constructed. Comparing regional, cell type/state-specific transcriptomic signatures using visual and quantitative decisions of an individual mouse with norms was performed. This analysis method allowed for the individuation-based evaluation of animals for their behavior correlates. We were able to obtain and reproduce a wide variety of behavior metrics, which is in this study included the alternation score on the Y-maze; the Y-maze alternation scores of 7-month-old wild-type mice ranged widely as well as those of 5xFAD mice, but those of 8.5-month-old wild-type mice converged with smaller variation to lower values, meaning commonly poorer performance at this age even in wild-type mice. After anti-CD4 antibody treatment, the variation was sustained with a slight improvement in their average scores (Fig. 4 B). After NK cell supplement treatment, variation was also sustained, with slight improvement in their average scores (Fig. 4 B). We assumed that these behavior variabilities are the keystone for proving the feasibility of tentative novel immunomodulatory treatments and that we would find that the mouse behavior scores concord with the transcriptomic signature [ 57 ]. NK cell-treated 5xFAD mice with higher Y-maze alternation scores definitely showed that their amygdala GABAergic Sst neuronal subtypes decreased in intensity (Fig. 5 A). This decrease (or increase, if any) did not prove the efficacy of NK cell supplement treatment on 5xFAD mice but definitely disclosed that transcriptomics of the neuronal subtype of that region were correlated with the degree of behavior impairment. More importantly, this meant that many other neuronal subtypes, other homeostatic or reactive glial cells and their subtypes, did not show any change in intensity over all the regions examined on these sections despite the improved behavior score. Anti-CD4 antibody treatment recapitulated only a slight decrease in specific immune cell signatures in the white matter, but beyond this finding, no other discovery of drug-responsive transcriptomic changes in any region or in any cell type or cell state was found. This was even observed on individual interpretations both visually and quantitatively for each mouse (Fig. 5 B and Supplementary Fig. 21 ). We could say that anti-CD4 immunoglobulins did not affect the transcriptomic signatures of major brain cells (on this single coronal section), and this was also the case with rare immune cells. Due to the lack of Y-maze score measures of the anti-CD4 antibody-treated wild-type and 5xFAD mice, behavior correlation could not be reported here.
The interpretation of rare immune cell signatures for the localization of immune cells presented different challenges from major brain cells. First, due to the intrinsic limitations of Visium, rare cell transcripts may not be well captured compared to the abundant cell type. Additionally, Visium captures a mixture of transcripts from multiple cells and lacks single-cell resolution. Therefore, it relies on cell type abundance estimation tools, which may be less reliable than image-based ST methods that capture transcript expression at the single-cell level. Nevertheless, we attempted to overcome the limitations with several strategies. The first was to remove the background effects of major brain cells. Homeostatic and reactive microglia and their coexpressed transcriptomes between microglia and infiltrating monocytes were the major challenge but were easy to remove, and astrocytes and oligodendrocytes followed by reactive glial cells expressed the same/similar transcriptomes. Double-checking the unique transcriptomes and their combinations was attempted with the data by Ximerakis et al. [ 64 ] and Schafflick et al. [ 68 ]. based on brain tissue studies. The study by Schafflick et al. [ 68 ]. used cells sorted by FACS for CD45 (gene Ptprc ) positivity and thus we were unable to remove the coexpressed transcriptomes of ribosomal protein transcriptomes for lymphoid cells, which if removed, would have enabled correct classification of myeloid and lymphoid cells among major brain cells in terms of intensity and distribution. Nevertheless, visual/manual curation by surveying individual transcriptomes helped to remove absurdly intense and unrealistically distributed transcriptomic signatures. When we used only the data of Schafflick et al. [ 68 ]. , we could not correct the inappropriate signature for B and T-cell compartments even after NSForest application to their data. The data came to look realistic after we adopted cross-tissue data and visual curation upon the two reports by Eraslan et al. [ 69 ] and Dominguez Conde et al. [ 71 ]. DEG data with an arbitrary threshold of 2.0 higher or -2.0 lower log fold change (LFC) for MHC + infiltrating immune macrophages (Mϕs) or LYVE + infiltrating integrity Mϕs produced 200 or more or 100 or more transcriptomes, respectively. We needed to remove, upon visual curation, 30% or 20% of transcriptomes to annotate the infiltrating monocyte-derived Mϕ. Infiltrating Mϕ and border-associated Mϕ [ 67 , 68 ] should have been differentiated but this was not possible due to the lack of clear distinction between the two cell types in the literature and the sparsity of the cells of both types. Tissue-resident and effector memory cells were traced with the transcriptomic signature by Dominguez Conde et al. [ 71 ]. As these authors included a variety of tissues (unfortunately, brain was not included) and stromal tissue specificity was considered a possible confounder in common for every tissue and thus, as expected, they yielded the signature for three compartments of T/ILC, B-cell and myeloid compartments. Of course, the types/subtypes of classically well-known immune cells belonging to these three compartments represented well the rare immune cells that would have originated from the bone marrow. We found differences in the intensity and distribution of the three compartments in the brain sections between 5xFAD mice and wild-type mice (Fig. 3 ). Drug/treatment effects should have been disclosed with this comparison, but we simply state that further investigation is warranted with a larger number of mice to avoid confounding factors which may hide or spuriously render probable false-negative/positive results regarding the effect of any tentative immunomodulatory treatments (Supplementary Tables 5 and 6 ).
The ultimate objective of using ST brain imaging with its visual and quantitative analysis is to convincingly designate the target cells, either major or rare, with regional localization; this can be for either brain parenchymal/stromal or rare immune cells, either resident or infiltrating immune cells and their homeostatic/reactive states, and target genes with significant contributions to pathologic changes in cells/regional tissues and their response to effective or ineffective treatments. More importantly, we could be sure that the unfound cells and transcriptomes were innocent, meaning that they were not affected by the test trial of a novel immunomodulatory therapy. For neuroimmune interactions during the disease process or in response to disease modifying drugs, we now know that the skull BM communicates with the dural sinus and peri-sinus regions, dural lymphatics as well as across ABC and CSF and thus perivascular spaces and ISF; communication can also take a totally different and unique route via the capillary endothelium and stroma of the choroid plexus, and choroid epithelium despite its tight junctions as well as the brain blood vessels’ microvascular endothelium despite its tight junctions. Once immune cells from the three compartments of T/ILCs, B cells and myeloid cells infiltrate the brain parenchyma, dynamically changing along the aging or disease process (in 5xFAD or P301L mice), they can respond to systemic immunomodulatory drug treatment directly or at least indirectly. The abundance of T cells (average 4/mm3) relative to neurons (90,000/mm3) or microglia (6,500/mm3) suggests that a few immune cells could change the response of major brain cells by significantly changing the transcriptomic signature of major brain cells. How the signals are transferred and/or translated from systemically administered anti-CD4 immunoglobulins or NK cell supplements should be investigated further. In this study, the study scheme and analysis methods were proposed to be applied to use ST brain imaging to investigate the impact of novel tentative disease-modifying treatments on neurodegenerative diseases and to elucidate whether regional brain cell-type/state-specific changes in the entire transcriptome per spot/region/cells of the brain or immune system would respond. The comprehensiveness and resolution of the results will be much improved with more novel technology [ 54 , 55 ] that will be available soon in many institutions, such as Visium methods [ 47 ]. Accordingly, methodology for analyzing spatial transcriptomics can be incorporated into high-resolution ST technologies to determine changes in cell types and abundance of rare immune cells with greater confidence.
Materials and methods
Ad models at different ages.
Three-month- and 7.5-month-old male 5xFAD mice (Tg6799; on a C57BL/6-SJL background) containing five FAD mutations in human APP (the Swedish mutation, K670N/M671L; the Florida mutation, I716V; and the London mutation, V717I; and the PS1 mutations M146L/L286V) and wild-type mice were used for spatial transcriptomic brain imaging data. Six- and seventeen-month-old male tau P301L mice (MAPT P301L mutations; on an FVB/N background) were used. Mice of all strains were raised in a laboratory cage with controlled temperature and humidity and on a 12 h light-dark cycle with free access to food and water. All experimental protocols and animal usage were approved (SNU-181018-6, SNU-190221-1-5) by the Institutional Animal Care and Use Committee (IACUC) at Seoul National University. All animals were handled in accordance with the Animal Research: Reporting of in vivo Experiments (ARRIVE) guidelines ( https://arriveguidelines.org ). Details are in Supplementary Notes.
Peripheral CD4 T-cell blockade in the 5xFAD AD model
Anti-CD4 antibody (0.5 mg/mouse; Bio X Cell) was intravenously injected into 6.5-month-old 5xFAD and wild-type mice according to group. Samples of different tissues were obtained after a month. Coronal sections of brain samples were used for spatial transcriptomic data acquisition.
Administration of NK cell supplement in the 5xFAD AD model
NK cells were expanded for 7 days after the isolation of NK cells from BALB/c mouse spleens. NK cells (2 × 10 6 cells/mouse in saline) were intravenously administered once a week for a total of five times to 6.5-month-old 5xFAD and wild-type mice. Brain samples were obtained after five weeks and used for spatial transcriptomic data.
Spatial gene expression library construction
Mice were anesthetized with isoflurane inhalation and perfused intracardially with cold DPBS (Dulbecco’s Phosphate-Buffered Saline; Gibco). Then, whole brains were removed. Brain hemispheres were prepared in frozen blocks using OCT compound (Sakura) and cryosectioned into 10 μm coronal and sagittal sections. According to the manufacturer’s protocols using Visium Spatial Tissue Optimization slides (10X Genomics), the permeabilization time was optimized to 12 min. The brain sections were methanol-fixed, hematoxylin and eosin (H&E)-stained and imaged on a TissueFAXS PLUS (TissueGenostics). The slides were merged into a picture of the whole brain using TissueFAXS imaging software. Then, the sections were permeabilized and processed to obtain cDNA Visium Spatial Gene Expression libraries according to the manufacturer’s protocol. To verify the size of PCR-enriched fragments, the template size distribution was checked using a high-sensitivity DNA assay (Agilent Technologies 2100 Bioanalyzer).
Generation of the count matrix
The libraries were sequenced using HiSeqXten (Illumina) with a read length of 28 bp for read 1 (Spatial Barcode and UMI), 10 bp index read (i7 index), 10 bp index read (i5 index), and 90 bp for read 2 (RNA read). Raw FASTQ data and H&E images were processed by the Space Ranger v1.1.0 (10X Genomics) pipeline for the gene expression analysis of Visium Spatial Gene Expression library data. Illumina base call files from the Illumina sequencing instrument were converted to FASTQ format for each sample using the ‘mkfastq’ command. Visium spatial expression libraries were analyzed with the ‘count’ command. Image alignment to predefined spots was performed using the fiducial alignment grid of the tissue image to determine the orientation and position of the input image. Sequencing reads were aligned to the mm10 reference genome (mm10-2020-A) using STAR (v2.5.1b) aligner. Gene expression profiling in each spot was performed with unique molecular identifier (UMI) and 10X barcode information.
Integration and spot clustering
A total of 63 spatial transcriptomic datasets, including brain tissue from wild-type and 5xFAD mice, with 32,885 genes in common were integrated and analyzed. The generated gene counts were normalized using ‘LogNormalize’ methods with a scale factor of 10,000. The top highly variable genes (HVGs; n = 2,000) in each tissue slide were identified using the variance stabilizing transformation (vst) method. The 2000 integration genes across all slides were then selected by ranking the genes by the number of slides in which they are variable in and their median rank of variability across the slides. For each slide, the log-normalized count matrix for the selected genes was scaled and the total RNA counts in each spot was regressed to remove the influence of the total count in the integration process. Principal component analysis (PCA) was performed for dimensionality reduction. Integration was performed for multiple spatial datasets prior to spot clustering to remove the batch effect. To flexibly integrate a large number of slides with both coronal and sagittal sections, reciprocal PCA (RPCA) was used to discover a set of anchors between the datasets, and normal (wild-type) mice were used as a reference during integration. The anchors were utilized to correct the count matrix in each spatial spot. The corrected counts were then scaled and PCA was performed. For spot clustering, a shared nearest neighbor (SNN) graph was constructed and graph-based clustering was performed using the Louvain algorithm. The resulting spot clusters were visualized using two different approaches: spatially mapped to the tissue based on spatial barcodes, or plotted in 2-dimensional space using Uniform Manifold Approximation and Projection (UMAP). The optimal resolution of the spot clusters was determined by decreasing the resolution value and visually examining the appropriate granularity of the spatial clusters that corresponded well to the anatomical structure. The anatomical location of each cluster was visually determined by comparison with the Allen Mouse Brain Reference Atlas ( https://mouse.brain-map.org/static/atlas ). As a result, the resolution was set to 0.15 for subsequent analysis. All analyses were performed using the R package, Seurat (version 4.1.1) [ 74 ].
Differential gene expression analysis
MAST (Model-based Analysis of Single-cell Transcriptomics) was used to perform differential gene expression analysis [ 96 ]. MAST accounts for the bimodal distribution of counts in the spatial transcriptomics and uses a generalized linear model with the proportion of genes expressed in each spot as a covariate to model the normalized counts. Differentially expressed genes (DEGs) were extracted from the comparison of wild-type and 5xFAD mice in each spot cluster defining the anatomical region in the brain. The cutoff for significantly different genes was false discovery rate (FDR)-adjusted p < 0.05 and log FC > 0.25.
Overrepresentation analysis
Overrepresentation analysis was performed and the Gene Ontology (GO) biological process terms associated with DEGs were identified. The count ratio was defined as the ratio of the proportion of the genes constituting GO terms among the DEGs to the proportion of genes constituting GO terms among total genes. Statistical significance was calculated based on the hypergeometric model, and correction for multiple comparisons was performed using the Benjamini-Hochberg procedure. The dot plots for the significant GO terms were drawn by showing the number of overlapping genes between the DEGs and each GO term, the count ratio, and the adjusted p-values. Overrepresentation tests were performed using clusterProfiler [ 118 ], which supports statistical analysis and visualization of functional profiles for gene sets. The packages ‘enrichplot’ and ‘igraph’ were additionally used to visualize the results.
Marker panel selection and curation
To analyze the spatial patterns of major cell types and immune cell types, the panel of marker genes was constructed and curated for each cell type. For the cell types identified in studies not using scRNAseq, individual genes were determined based on reference papers. For the cell types defined by scRNAseq, Necessary and Sufficient Forest (NSForest) version 2 [ 59 ] was applied and signature genes for the cell type were determined based on the cell type annotation information. The NSForest algorithm scores genes according to binary expression profiles in a specific cell type compared to other cell types. Then, based on the random forest algorithm, the minimum gene set that best describes the given cell type was searched. After selecting signature genes based on NSForest, the gene sets were refined to exclude the genes that are highly expressed in major cell types. This is particularly important when the scRNAseq data represent a subpopulation of the cells in the brain, such as immune cell sorted datasets. As a validation process, the spatial expression of the selected marker genes was examined and the genes were excluded if they showed a non-specific distribution pattern for the cell type. As a final step, genes that were not present in our spatial transcriptomic data were excluded. The curated gene sets are listed in Supplementary Table 7 .
Comparison of cellular signatures across groups
After curation of the marker panel, a gene set that best represents a particular cell type, the signature score of each cell type was computed on the spatial transcriptomic data by utilizing the AddModuleScore function in Seurat [ 44 ] with default parameters. For each gene in the gene set, a fixed number of control genes with the same average expression level as the gene were randomly selected. The difference between the average expression of the gene set and that of the control gene sets was calculated and named the cell signature score. The score in each spot was spatially mapped to the tissue using the SpatialFeaturePlot function in Seurat, and the spatial distribution pattern was identified. The average of the signature scores in a given region of interest was calculated and the values were compared between groups using the Wilcoxon rank-sum test. Correction for multiple comparison was performed using the Bonferroni method. The cutoff for the adjusted p-value was 0.05.
Cell type deconvolution analysis
The cell type distribution represented by the cell signature scores was compared to that derived by the cell type deconvolution method, CellDART [ 46 ]. CellDART first trains a model to extract cell type proportions from the synthetic mixture of cells generated from the reference scRNAseq dataset, and then adapts the model to predict the cell type composition of the spot, which is a mixture of multiple cells. For the major brain cell types, snRNAseq datasets from mouse brain coronal slices [ 49 ] were used as a reference for predicting spatial cell distribution. However, in the case of immune cells, the majority of scRNAseq datasets are obtained after cell sorting strategies such as fluorescence-activated cell sorting (FACS), and there is a mismatch in cell type and composition between spatial and single-cell datasets. Therefore, the cell type deconvolution tool spSeudoMap was used to compensate for this discrepancy [ 50 ]. For lymphoid and myeloid brain cell types, scRNAseq samples from CNS border immune cells were used [ 119 ], and for microglia, scRNAseq samples from brain immune cells were used [ 89 ]. The distribution of representative cell types that showed significant differences between wild-type and 5xFAD mice was evaluated: homeostatic microglia, reactive microglia, macrophages, monocytes, dendritic cells, innate lymphoid cells, natural killer cells, and T cells. The cell type annotation information from the reference single-cell dataset was used for the deconvolution analysis. Default parameter values suggested in the user manual were applied for the analysis.
Statistical analysis
For the spatial transcriptomic data, plots in R were created either with the ggplot2 R package or Seurat modified by custom codes for data visualization. All p-values reported in this study were adjusted by FDR (for DE analysis using MAST) using Benjamini-Hochberg procedure or Bonferroni method (all other analyses). The p-values below 0.05 were considered statistically significant.
Development of an application to visualize and quantify ST datasets
An R shiny-based application named STquantool was developed to comprehensively analyze ST datasets to explore cell type- and cell state-specific regional changes in wild-type, 5xFAD, and treatment mouse models. The application allows users to easily load and integrate the multiple ST datasets and visualize the spatial expression of genes and cell type scores based on Seurat [ 74 ] and shiny running on R (ver. 4.1.1). One of the key features of STquantool is that it facilitates the curation of cell type-specific marker combinations by sorting out key genes based on the NSForest [ 59 ] algorithm and finalizing the markers by visually assessing the spatial expression patterns. As an adjunct, the cell type decomposition method CellDART [ 46 ] can be implemented to find the spatial distribution patterns of major cell types constituting brain tissues. Moreover, the spatial patterns of the cell scores and cell fraction can be quantified and statistically analyzed with STquantool. Finally, the gene-level transcriptomic alterations between the mouse groups can be explored by performing the DEG analysis provided in the application. Then, the functional implications of the selected genes can be represented by gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) terms [ 120 , 121 , 122 ]. The suggested platform was packaged and can be readily installed from GitHub ( https://github.com/bsungwoo/STquantool.git ).
Data availability
All data are available in the main text or the supplementary materials. Additionally, the Visium spatial transcriptomics datasets utilized in this research are accessible on the data repository ( https://zenodo.org/records/10404412 ). The spatial transcriptomics analytical pipeline, STquantool, is available for installation from GitHub at https://github.com/bsungwoo/STquantool.git
Brioschi S, Wang WL, Peng V, Wang M, Shchukina I, Greenberg ZJ et al. Heterogeneity of meningeal B cells reveals a lymphopoietic niche at the CNS borders. Science. 2021;373.
Cugurra A, Mamuladze T, Rustenhoven J, Dykstra T, Beroshvili G, Greenberg ZJ et al. Skull and vertebral bone marrow are myeloid cell reservoirs for the meninges and CNS parenchyma. Science. 2021;373.
Cai R, Pan C, Ghasemigharagoz A, Todorov MI, Förstera B, Zhao S, et al. Panoptic imaging of transparent mice reveals whole-body neuronal projections and skull-meninges connections. Nat Neurosci. 2019;22:317–27.
Article CAS PubMed Google Scholar
Shibata-Germanos S, Goodman JR, Grieg A, Trivedi CA, Benson BC, Foti SC, et al. Structural and functional conservation of non-lumenized lymphatic endothelial cells in the mammalian leptomeninges. Acta Neuropathol. 2020;139:383–401.
Kutomi O, Takeda S. Identification of lymphatic endothelium in cranial arachnoid granulation-like dural gap. Microscopy (Oxf). 2020;69:391–400.
Louveau A, Smirnov I, Keyes TJ, Eccles JD, Rouhani SJ, Peske JD, et al. Structural and functional features of central nervous system lymphatic vessels. Nature. 2015;523:337–41.
Article CAS PubMed PubMed Central Google Scholar
Aspelund A, Antila S, Proulx ST, Karlsen TV, Karaman S, Detmar M, et al. A dural lymphatic vascular system that drains brain interstitial fluid and macromolecules. J Exp Med. 2015;212:991–9.
Bedussi B, van der Wel NN, de Vos J, van Veen H, Siebes M, VanBavel E, et al. Paravascular channels, cisterns, and the subarachnoid space in the rat brain: a single compartment with preferential pathways. J Cereb Blood Flow Metab. 2017;37:1374–85.
Article PubMed Google Scholar
Da Mesquita S, Louveau A, Vaccari A, Smirnov I, Cornelison RC, Kingsmore KM, et al. Functional aspects of meningeal lymphatics in ageing and Alzheimer’s disease. Nature. 2018;560:185–91.
Article PubMed PubMed Central Google Scholar
Ahn JH, Cho H, Kim JH, Kim SH, Ham JS, Park I, et al. Meningeal lymphatic vessels at the skull base drain cerebrospinal fluid. Nature. 2019;572:62–6.
Frederick N, Louveau A. Meningeal lymphatics, immunity and neuroinflammation. Curr Opin Neurobiol. 2020;62:41–7.
Papadopoulos Z, Herz J, Kipnis J. Meningeal lymphatics: from anatomy to Central Nervous System Immune Surveillance. J Immunol. 2020;204:286–93.
Alves de Lima K, Rustenhoven J, Kipnis J. Meningeal immunity and its function in maintenance of the Central Nervous System in Health and Disease. Annu Rev Immunol. 2020;38:597–620.
Hsu M, Sandor M, Fabry Z. Current concepts on communication between the central nervous system and peripheral immunity via lymphatics: what roles do lymphatics play in brain and spinal cord disease pathogenesis? Biol Futur. 2021;72:45–60.
Solár P, Zamani A, Kubíčková L, Dubový P, Joukal M. Choroid plexus and the blood-cerebrospinal fluid barrier in disease. Fluids Barriers CNS. 2020;17:35.
Lun MP, Monuki ES, Lehtinen MK. Development and functions of the choroid plexus-cerebrospinal fluid system. Nat Rev Neurosci. 2015;16:445–57.
Damkier H, Praetorius J. Structure of the Mammalian Choroid Plexus. In: Role of the Choroid Plexus in Health and Disease Edited by Praetorius J, Blazer-Yost B, Damkier H. New York, NY: Springer US; 2020: 1–33.
Fame RM, Lehtinen MK. Emergence and developmental roles of the Cerebrospinal Fluid System. Dev Cell. 2020;52:261–75.
Dani N, Herbst RH, McCabe C, Green GS, Kaiser K, Head JP, et al. A cellular and spatial map of the choroid plexus across brain ventricles and ages. Cell. 2021;184:3056–74.
Agarwal N, Carare RO. Cerebral vessels: an overview of anatomy, physiology, and role in the drainage of fluids and solutes. Front Neurol. 2020;11:611485.
Ross JM, Kim C, Allen D, Crouch EE, Narsinh K, Cooke DL, et al. Expanding Cell Divers Brain Vasculature Front Physiol. 2020;11:600767.
Google Scholar
Kalucka J, de Rooij L, Goveia J, Rohlenova K, Dumas SJ, Meta E, et al. Single-cell transcriptome atlas of murine endothelial cells. Cell. 2020;180:764–79.
Vanlandewijck M, He L, Mäe MA, Andrae J, Ando K, Del Gaudio F, et al. A molecular atlas of cell types and zonation in the brain vasculature. Nature. 2018;554:475–80.
Dixon GA, Pérez CA. Multiple sclerosis and the Choroid Plexus: emerging concepts of Disease Immunopathophysiology. Pediatr Neurol. 2020;103:65–75.
Stock AD, Der E, Gelb S, Huang M, Weidenheim K, Ben-Zvi A, et al. Tertiary lymphoid structures in the choroid plexus in neuropsychiatric lupus. JCI Insight. 2019;4:e124203.
Ma Q, Decker Y, Müller A, Ineichen BV, Proulx ST. Clearance of cerebrospinal fluid from the sacral spine through lymphatic vessels. J Exp Med. 2019;216:2492–502.
Jacob L, Boisserand L, Geraldo BS, de Brito Neto LMH. Anatomy and function of the vertebral column lymphatic network in mice. Nat Commun. 2019;10:4594.
Petrova TV, Koh GY. Biological functions of lymphatic vessels. Science. 2020;369:eaax4063.
Lee EJ, Choi Y, Park EJ, Lee DS. Lymphatic dysfunction sustains memory impairment despite Abeta reduction in an Alzheimer’s disease model. Immunity & Ageing; 2022. (in revision).
Lee EJ. Investigation of the spatial transcriptomic signatures and therapeutic mode of action in an Alzheimer’s disease model. Seoul National University (Thesis). 2022.
Huang Q, Belz GT. Parallel worlds of the adaptive and innate immune cell networks. Curr Opin Immunol. 2019;58:53–9.
Unger MS, Li E, Scharnagl L, Poupardin R, Altendorfer B, Mrowetz H, et al. CD8(+) T-cells infiltrate Alzheimer’s disease brains and regulate neuronal- and synapse-related gene expression in APP-PS1 transgenic mice. Brain Behav Immun. 2020;89:67–86.
Laurent C, Dorothée G, Hunot S, Martin E, Monnet Y, Duchamp M, et al. Hippocampal T cell infiltration promotes neuroinflammation and cognitive decline in a mouse model of tauopathy. Brain. 2017;140:184–200.
Faridar A, Thome AD, Zhao W, Thonhoff JR, Beers DR, Pascual B, et al. Restoring regulatory T-cell dysfunction in Alzheimer’s disease through ex vivo expansion. Brain Commun. 2020;2:fcaa112.
Dansokho C, Ait Ahmed D, Aid S, Toly-Ndour C, Chaigneau T, Calle V, et al. Regulatory T cells delay disease progression in Alzheimer-like pathology. Brain. 2016;139:1237–51.
Gingele S, Pul R, Sardari M, Borbor M, Henkel F, Moellenkamp TM, et al. FoxP3 deficiency causes no inflammation or neurodegeneration in the murine brain. J Neuroimmunol. 2020;342:577216.
Ito M, Komai K, Mise-Omata S, Iizuka-Koga M, Noguchi Y, Kondo T, et al. Brain regulatory T cells suppress astrogliosis and potentiate neurological recovery. Nature. 2019;565:246–50.
Krämer TJ, Hack N, Brühl TJ, Menzel L, Hummel R, Griemert EV, et al. Depletion of regulatory T cells increases T cell brain infiltration, reactive astrogliosis, and interferon-γ gene expression in acute experimental traumatic brain injury. J Neuroinflammation. 2019;16:163.
Fisher Y, Strominger I, Biton S, Nemirovsky A, Baron R, Monsonego A. Th1 polarization of T cells injected into the cerebrospinal fluid induces brain immunosurveillance. J Immunol. 2014;192:92–102.
Martinez B, Peplow PV. Amelioration of Alzheimer’s disease pathology and cognitive deficits by immunomodulatory agents in animal models of Alzheimer’s disease. Neural Regen Res. 2019;14:1158–76.
Mittal K, Eremenko E, Berner O, Elyahu Y, Strominger I, Apelblat D, et al. CD4 T cells induce a subset of MHCII-Expressing microglia that attenuates Alzheimer Pathology. iScience. 2019;16:298–311.
Raha-Chowdhury R, Henderson JW, Raha AA, Vuono R, Bickerton A, Jones E, et al. Choroid Plexus acts as Gatekeeper for TREM2, abnormal Accumulation of ApoE, and Fibrillary Tau in Alzheimer’s Disease and in Down Syndrome Dementia. J Alzheimers Dis. 2019;69:91–109.
Satija R, Farrell JA, Gennert D, Schier AF, Regev A. Spatial reconstruction of single-cell gene expression data. Nat Biotechnol. 2015;33:495–502.
Tirosh I, Izar B, Prakadan SM, Wadsworth MH 2nd, Treacy D, Trombetta JJ, et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science. 2016;352:189–96.
Butler A, Hoffman P, Smibert P, Papalexi E, Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol. 2018;36:411–20.
Bae S, Na KJ, Koh J, Lee DS, Choi H, Kim YT. CellDART: cell type inference by domain adaptation of single-cell and spatial transcriptomic data. Nucleic Acids Res. 2022;50:e57.
Ståhl PL, Salmén F, Vickovic S, Lundmark A, Navarro JF, Magnusson J, et al. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science. 2016;353:78–82.
Cable DM, Murray E, Zou LS, Goeva A, Macosko EZ, Chen F, et al. Robust decomposition of cell type mixtures in spatial transcriptomics. Nat Biotechnol. 2022;40:517–26.
Kleshchevnikov V, Shmatko A, Dann E, Aivazidis A, King HW, Li T, et al. Cell2location maps fine-grained cell types in spatial transcriptomics. Nat Biotechnol. 2022;40:661–71.
Bae S, Choi H, Lee DS, spSeudoMap. Cell type mapping of spatial transcriptomics using unmatched single-cell RNA-seq data. Genome Med. 2023;15:19.
Vickovic S, Eraslan G, Salmén F, Klughammer J, Stenbeck L, Schapiro D, et al. High-definition spatial transcriptomics for in situ tissue profiling. Nat Methods. 2019;16:987–90.
Stickels RR, Murray E, Kumar P, Li J, Marshall JL, Di Bella DJ, et al. Highly sensitive spatial transcriptomics at near-cellular resolution with Slide-seqV2. Nat Biotechnol. 2021;39:313–9.
Cho CS, Xi J, Si Y, Park SR, Hsu JE, Kim M, et al. Microscopic examination of spatial transcriptome using seq-scope. Cell. 2021;184:3559–72.
Chen A, Liao S, Cheng M, Ma K, Wu L, Lai Y, et al. Spatiotemporal transcriptomic atlas of mouse organogenesis using DNA nanoball-patterned arrays. Cell. 2022;185:1777–92.
Chen A, Sun Y, Lei Y, Li C, Liao S, Liang Z, et al. Single-cell spatial transcriptome reveals cell-type organization in the macaque cortex. Cell. 2023;186:3726–43.
Wei X, Fu S, Li H, Liu Y, Wang S, Feng W, et al. Single-cell stereo-seq reveals induced progenitor cells involved in axolotl brain regeneration. Science. 2022;377:eabp9444.
Bugeon S, Duffield J, Dipoppa M, Ritoux A, Prankerd I, Nicoloutsopoulos D, et al. A transcriptomic axis predicts state modulation of cortical interneurons. Nature. 2022;607:330–8.
Zeng H, de Vries SEJ. A gene-expression axis defines neuron behaviour. Nature. 2022;607:243–4.
Aevermann B, Zhang Y, Novotny M, Keshk M, Bakken T, Miller J, et al. A machine learning method for the discovery of minimum marker gene combinations for cell type identification from single-cell RNA sequencing. Genome Res. 2021;31:1767–80.
Nelson ME, Riva SG, Cvejic A. SMaSH: a scalable, general marker gene identification framework for single-cell RNA-sequencing. BMC Bioinformatics. 2022;23:328.
Vargo AHS, Gilbert AC. A rank-based marker selection method for high throughput scRNA-seq data. BMC Bioinformatics. 2020;21:477.
Delaney C, Schnell A, Cammarata LV, Yao-Smith A, Regev A, Kuchroo VK, et al. Combinatorial prediction of marker panels from single-cell transcriptomic data. Mol Syst Biol. 2019;15:e9005.
Armingol E, Officer A, Harismendy O, Lewis NE. Deciphering cell-cell interactions and communication from gene expression. Nat Rev Genet. 2021;22:71–88.
Ximerakis M, Lipnick SL, Innes BT, Simmons SK, Adiconis X, Dionne D, et al. Single-cell transcriptomic profiling of the aging mouse brain. Nat Neurosci. 2019;22:1696–708.
Saunders A, Macosko EZ, Wysoker A, Goldman M, Krienen FM, de Rivera H, et al. Molecular diversity and specializations among the cells of the adult mouse brain. Cell. 2018;174:1015–30.
Marques S, Zeisel A, Codeluppi S, van Bruggen D, Mendanha Falcao A, Xiao L, et al. Oligodendrocyte heterogeneity in the mouse juvenile and adult central nervous system. Science. 2016;352:1326–9.
Van Hove H, Martens L, Scheyltjens I, De Vlaminck K, Pombo Antunes AR, De Prijck S, et al. A single-cell atlas of mouse brain macrophages reveals unique transcriptional identities shaped by ontogeny and tissue environment. Nat Neurosci. 2019;22:1021–35.
Schafflick D, Wolbert J, Heming M, Thomas C, Hartlehnert M, Börsch AL, et al. Single-cell profiling of CNS border compartment leukocytes reveals that B cells and their progenitors reside in non-diseased meninges. Nat Neurosci. 2021;24:1225–34.
Eraslan G, Drokhlyansky E, Anand S, Fiskin E, Subramanian A, Slyper M, et al. Single-nucleus cross-tissue molecular reference maps toward understanding disease gene function. Science. 2022;376:eabl4290.
Miragaia RJ, Gomes T, Chomka A, Jardine L, Riedel A, Hegazy AN, et al. Single-Cell Transcriptomics of Regulatory T Cells reveals trajectories of tissue adaptation. Immunity. 2019;50:493–504.
Domínguez Conde C, Xu C, Jarvis LB, Rainbow DB, Wells SB, Gomes T, et al. Cross-tissue immune cell analysis reveals tissue-specific features in humans. Science. 2022;376:eabl5197.
Kim DG, Lee JI, Lee DS, Lee MC, Choi KS, Han DH. 99mTc-HMPAO labeled leukocyte SPECT in intracranial lesions. Surg Neurol. 1995;44:338–45.
Tappan SJ, Eastwood BS, O’Connor N, Wang Q, Ng L, Feng D, et al. Automatic navigation system for the mouse brain. J Comp Neurol. 2019;527:2200–11.
Hao Y, Hao S, Andersen-Nissen E, Mauck WM 3rd, Zheng S, Butler A, et al. Integrated analysis of multimodal single-cell data. Cell. 2021;184:3573–e35873529.
Hodge RD, Bakken TE, Miller JA, Smith KA, Barkan ER, Graybuck LT, et al. Conserved cell types with divergent features in human versus mouse cortex. Nature. 2019;573:61–8.
Werkman IL, Dubbelaar ML, van der Vlies P, de Boer-Bergsma JJ, Eggen BJL, Baron W. Transcriptional heterogeneity between primary adult grey and white matter astrocytes underlie differences in modulation of in vitro myelination. J Neuroinflammation. 2020;17:373.
A multimodal cell. Census and atlas of the mammalian primary motor cortex. Nature. 2021;598:86–102.
Article Google Scholar
Tasic B, Yao Z, Graybuck LT, Smith KA, Nguyen TN, Bertagnolli D, et al. Shared and distinct transcriptomic cell types across neocortical areas. Nature. 2018;563:72–8.
Zeng H, Sanes JR. Neuronal cell-type classification: challenges, opportunities and the path forward. Nat Rev Neurosci. 2017;18:530–46.
Tasic B, Menon V, Nguyen TN, Kim TK, Jarsky T, Yao Z, et al. Adult mouse cortical cell taxonomy revealed by single cell transcriptomics. Nat Neurosci. 2016;19:335–46.
Yuste R, Hawrylycz M, Aalling N, Aguilar-Valles A, Arendt D, Armañanzas R, et al. A community-based transcriptomics classification and nomenclature of neocortical cell types. Nat Neurosci. 2020;23:1456–68.
Yao Z, van Velthoven CTJ, Nguyen TN, Goldy J, Sedeno-Cortes AE, Baftizadeh F, et al. A taxonomy of transcriptomic cell types across the isocortex and hippocampal formation. Cell. 2021;184:3222–e32413226.
Bayraktar OA, Bartels T, Holmqvist S, Kleshchevnikov V, Martirosyan A, Polioudakis D, et al. Astrocyte layers in the mammalian cerebral cortex revealed by a single-cell in situ transcriptomic map. Nat Neurosci. 2020;23:500–9.
Escartin C, Galea E, Lakatos A, O’Callaghan JP, Petzold GC, Serrano-Pozo A, et al. Reactive astrocyte nomenclature, definitions, and future directions. Nat Neurosci. 2021;24:312–25.
Habib N, McCabe C, Medina S, Varshavsky M, Kitsberg D, Dvir-Szternfeld R, et al. Disease-associated astrocytes in Alzheimer’s disease and aging. Nat Neurosci. 2020;23:701–6.
Ioannou MS, Jackson J, Sheu SH, Chang CL, Weigel AV, Liu H, et al. Neuron-astrocyte metabolic coupling protects against Activity-Induced fatty acid toxicity. Cell. 2019;177:1522–35.
Chamling X, Kallman A, Fang W, Berlinicke CA, Mertz JL, Devkota P, et al. Single-cell transcriptomic reveals molecular diversity and developmental heterogeneity of human stem cell-derived oligodendrocyte lineage cells. Nat Commun. 2021;12:652.
Krasemann S, Madore C, Cialic R, Baufeld C, Calcagno N, El Fatimy R, et al. The TREM2-APOE pathway drives the Transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity. 2017;47:566–81.
Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell. 2017;169:1276–e12901217.
Marschallinger J, Iram T, Zardeneta M, Lee SE, Lehallier B, Haney MS, et al. Lipid-droplet-accumulating microglia represent a dysfunctional and proinflammatory state in the aging brain. Nat Neurosci. 2020;23:194–208.
Friedman BA, Srinivasan K, Ayalon G, Meilandt WJ, Lin H, Huntley MA, et al. Diverse brain myeloid expression profiles reveal distinct microglial activation States and aspects of Alzheimer’s Disease Not Evident in Mouse models. Cell Rep. 2018;22:832–47.
Uriarte Huarte O, Richart L, Mittelbronn M, Michelucci A. Microglia in Health and Disease: the strength to be diverse and reactive. Front Cell Neurosci. 2021;15:660523.
Grubman A, Choo XY, Chew G, Ouyang JF, Sun G, Croft NP, et al. Transcriptional signature in microglia associated with Aβ plaque phagocytosis. Nat Commun. 2021;12:3015.
Pan J, Ma N, Yu B, Zhang W, Wan J. Transcriptomic profiling of microglia and astrocytes throughout aging. J Neuroinflammation. 2020;17:97.
Choi H, Lee EJ, Shin JS, Kim H, Bae S, Choi Y et al. Spatiotemporal characterization of glial cell activation in an Alzheimer’s disease model by spatially resolved transcriptomics. Exp Mol Med. 2023.
Finak G, McDavid A, Yajima M, Deng J, Gersuk V, Shalek AK, et al. MAST: a flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biol. 2015;16:278.
Locatelli G, Engelhardt B. Microglia get a little help from Th-eir friends. Immunity. 2020;53:484–6.
Erö C, Gewaltig MO, Keller D, Markram H. A cell atlas for the mouse brain. Front Neuroinform. 2018;12:84.
Keller D, Erö C, Markram H. Cell densities in the mouse brain: a systematic review. Front Neuroanat. 2018;12:83.
Mundt S, Mrdjen D, Utz SG, Greter M, Schreiner B, Becher B. Conventional DCs sample and present myelin antigens in the healthy CNS and allow parenchymal T cell entry to initiate neuroinflammation. Sci Immunol. 2019;4:eaau8380.
Scheurer L, Das Gupta RR, Saebisch A, Grampp T, Benke D, Zeilhofer HU et al. Expression of immunoglobulin constant domain genes in neurons of the mouse central nervous system. Life Sci Alliance. 2021;4.
Fang F, Cao W, Zhu W, Lam N, Li L, Gaddam S, et al. The cell-surface 5′-nucleotidase CD73 defines a functional T memory cell subset that declines with age. Cell Rep. 2021;37:109981.
Yang C, Siebert JR, Burns R, Gerbec ZJ, Bonacci B, Rymaszewski A, et al. Heterogeneity of human bone marrow and blood natural killer cells defined by single-cell transcriptome. Nat Commun. 2019;10:3931.
Crinier A, Milpied P, Escalière B, Piperoglou C, Galluso J, Balsamo A, et al. High-dimensional single-cell analysis identifies organ-specific signatures and conserved NK Cell subsets in humans and mice. Immunity. 2018;49:971–86.
Kim JS, Kolesnikov M, Peled-Hajaj S, Scheyltjens I, Xia Y, Trzebanski S, et al. A binary cre Transgenic Approach dissects Microglia and CNS border-Associated macrophages. Immunity. 2021;54:176–90.
Song YH, Yoon J, Lee SH. The role of neuropeptide somatostatin in the brain and its application in treating neurological disorders. Exp Mol Med. 2021;53:328–38.
Davies P, Katzman R, Terry RD. Reduced somatostatin-like immunoreactivity in cerebral cortex from cases of Alzheimer disease and Alzheimer senile dementa. Nature. 1980;288:279–80.
Jordão MJC, Sankowski R, Brendecke SM, Sagar, Locatelli G, Tai YH, et al. Single-cell profiling identifies myeloid cell subsets with distinct fates during neuroinflammation. Science. 2019;363:eaat7554.
Masuda T, Sankowski R, Staszewski O, Bottcher C, Amann L, Sagar, et al. Spatial and temporal heterogeneity of mouse and human microglia at single-cell resolution. Nature. 2019;566:388–92.
Sankowski R, Böttcher C, Masuda T, Geirsdottir L, Sagar, Sindram E, et al. Mapping microglia states in the human brain through the integration of high-dimensional techniques. Nat Neurosci. 2019;22:2098–110.
Kierdorf K, Masuda T, Jordão MJC, Prinz M. Macrophages at CNS interfaces: ontogeny and function in health and disease. Nat Rev Neurosci. 2019;20:547–62.
Prinz M, Jung S, Priller J. Microglia Biology: one century of evolving concepts. Cell. 2019;179:292–311.
Masuda T, Amann L, Sankowski R, Staszewski O, Lenz M. Author correction: Novel Hexb-based tools for studying microglia in the CNS. Nat Immunol. 2020;21:1302.
Borst K, Prinz M. Deciphering the heterogeneity of myeloid cells during neuroinflammation in the single-cell era. Brain Pathol. 2020;30:1192–207.
Dogra P, Rancan C, Ma W, Toth M, Senda T, Carpenter DJ, et al. Tissue determinants of human NK Cell Development, function, and Residence. Cell. 2020;180:749–63.
Fulcher BD, Arnatkeviciute A, Fornito A. Overcoming false-positive gene-category enrichment in the analysis of spatially resolved transcriptomic brain atlas data. Nat Commun. 2021;12:2669.
Chai H, Diaz-Castro B, Shigetomi E, Monte E, Octeau JC, Yu X, et al. Neural circuit-specialized astrocytes: Transcriptomic, Proteomic, Morphological, and functional evidence. Neuron. 2017;95:531–49.
Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. Omics. 2012;16:284–7.
Posner DA, Lee CY, Portet A, Clatworthy MR. Humoral immunity at the brain borders in homeostasis. Curr Opin Immunol. 2022;76:102188.
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al. Gene Ontology: tool for the unification of biology. Nat Genet. 2000;25:25–9.
The Gene Ontology. Resource: enriching a GOld mine. Nucleic Acids Res. 2021;49:D325–34.
Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30.
Download references
Acknowledgements
We thank Joo Young Park for his technical assistance. The work was supported by grants from the National Research Foundation of Korea (NRF-2017M3C7A1048079, NRF- 2020R1A2C2101069, NRF- 2022R1A5A6000840).
This work was supported by National Research Foundation of Korea (NRF) Grants funded by the Korean Government (MSIP) (No. 2017M3C7A1048079, No. 2020R1A2C2101069 and No. 2017R1A5A1015626).
Author information
Authors and affiliations.
Department of Nuclear Medicine, Seoul National University College of Medicine, 101 Daehak-ro, Jongno-gu, Seoul, 03080, Republic of Korea
Eun Ji Lee, Minseok Suh, Hongyoon Choi, Yoori Choi, Sungwoo Bae & Dong Soo Lee
Department of Molecular Medicine and Biopharmaceutical Sciences, Graduate School of Convergence Science and Technology, Seoul National University, Seoul, Republic of Korea
Eun Ji Lee & Dong Soo Lee
Department of Nuclear Medicine, Seoul National University Hospital, Seoul, Republic of Korea
Minseok Suh, Hongyoon Choi, Sungwoo Bae & Dong Soo Lee
Institute of Radiation Medicine, Medical Research Center, Seoul National University, 103 Daehak-ro, Jongno-gu, Seoul, 03080, Republic of Korea
Minseok Suh, Sungwoo Bae & Dong Soo Lee
Cliniclal Research Institute, Seoul National University Hospital, Seoul, Republic of Korea
Research and Development Center, THERABEST Inc., Seocho-daero 40-gil, Seoul, 06657, Republic of Korea
Do Won Hwang
Medical Science and Engineering, School of Convergence Science and Technology, POSTECH, Pohang, Republic of Korea
Dong Soo Lee
You can also search for this author in PubMed Google Scholar
Contributions
EJL: Methodology: Investigation: Visualization: Writing; SB: Methodology: Investigation: Visualization: Revision; MS: Methodology: Investigation: Visualization; HC: Methodology: Investigation: Funding acquisition; YC: Methodology: Investigation; DWH: Methodology: Investigation; DSL: Conceptualization: Methodology: Investigation: Visualization: Funding acquisition: Project administration: Supervision: Writing – original draft: Writing – review & editing.
Corresponding authors
Correspondence to Sungwoo Bae or Dong Soo Lee .
Ethics declarations
Ethics approval and consent to participate.
All experimental protocols and animal usage were approved (SNU-181018-6, SNU-190221-1-5) by the Institutional Animal Care and Use Committee (IACUC) at Seoul National University. All animals were handled in accordance with the Animal Research: Reporting of in vivo Experiments (ARRIVE) guidelines ( https://arriveguidelines.org ). Details are in Supplementary Notes.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note.
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Material 1
Supplementary material 2, supplementary material 3, rights and permissions.
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/ . The Creative Commons Public Domain Dedication waiver ( http://creativecommons.org/publicdomain/zero/1.0/ ) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
Reprints and permissions
About this article
Cite this article.
Lee, E.J., Suh, M., Choi, H. et al. Spatial transcriptomic brain imaging reveals the effects of immunomodulation therapy on specific regional brain cells in a mouse dementia model. BMC Genomics 25 , 516 (2024). https://doi.org/10.1186/s12864-024-10434-8
Download citation
Received : 18 February 2024
Accepted : 20 May 2024
Published : 25 May 2024
DOI : https://doi.org/10.1186/s12864-024-10434-8
Share this article
Anyone you share the following link with will be able to read this content:
Sorry, a shareable link is not currently available for this article.
Provided by the Springer Nature SharedIt content-sharing initiative
- Spatial transcriptomics
- Brain imaging
- Cell type decomposition
- Cell state annotation
- Major brain cells
- Rare immune cells
- Immunomodulatory therapy
BMC Genomics
ISSN: 1471-2164
- Submission enquiries: [email protected]
- General enquiries: [email protected]
Log in using your username and password
- Search More Search for this keyword Advanced search
- Latest content
- Current issue
- BMJ Journals More You are viewing from: Google Indexer
You are here
- Online First
- Identification and validation of anti-protein arginine methyltransferase 5 (PRMT5) antibody as a novel biomarker for systemic sclerosis (SSc)
- Article Text
- Article info
- Citation Tools
- Rapid Responses
- Article metrics
- http://orcid.org/0000-0002-7650-1237 Minrui Liang 1 , 2 , 3 ,
- Lingbiao Wang 1 , 2 , 3 ,
- Xiaolong Tian 4 ,
- Kun Wang 5 , 6 ,
- Xiaoyi Zhu 4 ,
- Linlin Huang 1 , 2 , 3 ,
- Qing Li 5 , 6 ,
- Wenjing Ye 1 , 2 , 3 ,
- Chen Chen 1 , 2 , 7 ,
- Haihua Yang 8 ,
- Wanqing Wu 9 ,
- Xiangjun Chen 9 ,
- Xiaoxia Zhu 1 , 2 , 3 ,
- Yu Xue 1 , 2 , 3 ,
- Weiguo Wan 1 , 2 , 3 ,
- Yanling Wu 4 ,
- http://orcid.org/0000-0002-8634-0967 Liwei Lu 10 ,
- http://orcid.org/0000-0003-2765-0620 Jiucun Wang 11 ,
- Hejian Zou 1 , 2 , 3 ,
- Tianlei Ying 4 ,
- Feng Zhou 5 , 6
- 1 Department of Rheumatology , Huashan Hospital, Fudan University , Shanghai , China
- 2 Institute of Rheumatology, Immunology and Allergy , Fudan University , Shanghai , China
- 3 Huashan Rare Disease Center , Huashan Hospital, Fudan University , Shanghai , China
- 4 Key Laboratory of Medical Molecular Virology (MOE/NHC/CAMS) and Shanghai Institute of Infectious Disease and Biosecurity, Shanghai Frontiers Science Center of Pathogenic Microorganisms and Infection, Shanghai Engineering Research Center for Synthetic Immunology, School of Basic Medical Sciences , Fudan University , Shanghai , China
- 5 Shanghai Key Laboratory of Medical Epigenetics, International Co-laboratory of Medical Epigenetics and Metabolism, Ministry of Science and Technology, Institutes of Biomedical Sciences , Fudan University , Shanghai , China
- 6 Key Laboratory of Carcinogenesis and Cancer Invasion, Ministry of Education, Liver Cancer Institute, Zhongshan Hospital , Fudan University , Shanghai , China
- 7 Department of Emergency Medicine , Zhongshan Hospital, Fudan University , Shanghai , China
- 8 Department of Respiratory and Critical Care Medicine , Huashan Hospital, Fudan University , Shanghai , China
- 9 Department of Neurology , Huashan Hospital, Fudan University , Shanghai , China
- 10 Department of Pathology , The University of Hong Kong , Hong Kong , China
- 11 State Key Laboratory of Genetic Engineering, Collaborative Innovation Center for Genetics and Development, School of Life Sciences, and Human Phenome Institute , Fudan University , Shanghai , China
- Correspondence to Dr Minrui Liang, Department of Rheumatology, Institute of Rheumatology, Immunology and Allergy; Huashan Rare Disease Center, Huashan Hospital, Fudan University, Shanghai, China; mliang10{at}fudan.edu.cn ; Professor Tianlei Ying, Key Laboratory of Medical Molecular Virology (MOE/NHC/CAMS) and Shanghai Institute of Infectious Disease and Biosecurity, Shanghai Frontiers Science Center of Pathogenic Microorganisms and Infection, Shanghai Engineering Research Center for Synthetic Immunology, School of Basic Medical Sciences, Fudan University, Shanghai, China; tlying{at}fudan.edu.cn ; Professor Feng Zhou, Shanghai Key Laboratory of Medical Epigenetics, International Co-laboratory of Medical Epigenetics and Metabolism, Ministry of Science and Technology, Institutes of Biomedical Sciences, Fudan University, and Key Laboratory of Carcinogenesis and Cancer Invasion, Ministry of Education, Liver Cancer Institute, Zhongshan Hospital, Fudan University, Shanghai, China; zhou_feng{at}fudan.edu.cn
Objectives In the complex panorama of autoimmune diseases, the characterisation of pivotal contributing autoantibodies that are involved in disease progression remains challenging. This study aimed to employ a global antibody profiling strategy to identify novel antibodies and investigate their association with systemic sclerosis (SSc).
Methods We implemented this strategy by conducting immunoprecipitation (IP) following on-bead digestion with the sera of patients with SSc or healthy donors, using antigen pools derived from cell lysates. The enriched antigen-antibody complex was proceeded with mass spectrometry (MS)-based quantitative proteomics and over-represented by bioinformatics analysis. The candidate antibodies were then orthogonally validated in two independent groups of patients with SSc. Mice were immunised with the target antigen, which was subsequently evaluated by histological examination and RNA sequencing.
Results The IP-MS analysis, followed by validation in patients with SSc, revealed a significant elevation in anti-PRMT5 antibodies among patients with SSc. These antibodies exhibited robust diagnostic accuracy in distinguishing SSc from healthy controls and other autoimmune conditions, including systemic lupus erythematosus and Sjögren’s syndrome, with an area under the curve ranging from 0.900 to 0.988. The elevation of anti-PRMT5 antibodies was verified in a subsequent independent group with SSc using an additional method, microarray. Notably, 31.11% of patients with SSc exhibited seropositivity for anti-PRMT5 antibodies. Furthermore, the titres of anti-PRMT5 antibodies demonstrated a correlation with the progression or regression trajectory in SSc. PRMT5 immunisation displayed significant inflammation and fibrosis in both the skin and lungs of mice. This was concomitant with the upregulation of multiple proinflammatory and profibrotic pathways, thereby underscoring a potentially pivotal role of anti-PRMT5 antibodies in SSc.
Conclusions This study has identified anti-PRMT5 antibodies as a novel biomarker for SSc.
- Autoantibodies
- Autoimmune Diseases
- Scleroderma, Systemic
Data availability statement
Data are available upon reasonable request. All data relevant to the study are presented in the article or uploaded as supplementary information.
This is an open access article distributed in accordance with the Creative Commons Attribution 4.0 Unported (CC BY 4.0) license, which permits others to copy, redistribute, remix, transform and build upon this work for any purpose, provided the original work is properly cited, a link to the licence is given, and indication of whether changes were made. See: https://creativecommons.org/licenses/by/4.0/ .
https://doi.org/10.1136/ard-2024-225596
Statistics from Altmetric.com
Request permissions.
If you wish to reuse any or all of this article please use the link below which will take you to the Copyright Clearance Center’s RightsLink service. You will be able to get a quick price and instant permission to reuse the content in many different ways.
WHAT IS ALREADY KNOWN ON THIS TOPIC
The involvement of autoantibodies in the development of systemic sclerosis (SSc), particularly those not linked to well-defined autoantigens, remains largely unknown.
SSc-specific autoantibodies play a critical role in the diagnosis, differentiation and stratification of the disease.
Ongoing efforts are underway to discover novel autoantibodies using advanced techniques.
WHAT THIS STUDY ADDS
This study represents the initial identification and validation of anti-PRMT5 antibodies in two independent groups of patients with SSc.
Levels of anti-PRMT5 antibodies exhibited a correlation with the disease trajectory in SSc, serving as a predictive indicator for regression or progression in both skin and lung involvement.
Immunisation with recombinant protein PRMT5 induced SSc-like manifestations in mice, indicating a potentially pivotal role in SSc.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
Anti-PRMT5 antibodies manifested as a diagnostic and predictive marker for SSc.
The application of anti-PRMT5 antibodies may contribute to precisive disease monitoring and prognosis.
Introduction
Systemic sclerosis (SSc) is an autoimmune rheumatic disease, characteristic of autoimmunity with increased inflammatory burden, vasculopathy with extensive endothelial dysfunction and tissue fibrosis with fibroblast activation. 1–4 An abundance of autoantibodies detected in SSc and the close link with clinical outcomes indicate the potential involvement of autoantibodies and the breakdown of self-tolerance in the pathogenesis of SSc, thereby offering up important novel diagnostic and therapeutic opportunities of autoantibodies. 5 6 Nevertheless, the well-defined SSc autoantigens are ubiquitously expressed and play an essential role in physiological processes. Consequently, the strong association of specific SSc autoantibodies with clinical phenotypes raises intriguing questions regarding their roles in SSc pathogenesis: are these autoantibodies the drivers or merely incidental bystanders?
Antitopoisomerase antibodies (ATAs), anticentromere antibodies (ACAs) and anti-RNA polymerase III antibodies (ARAs), three types of antinuclear antibodies (ANAs), have been reported as the most prevalent SSc-specific antibodies. However, up to 11% of patients with SSc are negative for ANA, 7 and even as many as 17% of patients with SSc lack detectable levels of established SSc-specific antibodies. 8 This highlights the necessity to explore novel autoantibodies that are specific to SSc. There is a growing body of studies focusing on the identification of novel autoantibodies and the definition of their clinical implications, such as antiplatelet-derived growth factor receptor (PDGFR), antiangiotensin receptor type 1, antigephyrin and antieukaryotic initiation factor 2B antibodies. 9–14 Mechanistically, single-cell analysis reveals broad differences in cell cluster gene expression profiles, showing distinctions in clinical phenotypes and distinct skin score trajectories across autoantibody subgroups of diffuse cutaneous SSc (dcSSc). 15 However, further investigation is required to elucidate the precise pathomechanism of autoantibodies in SSc.
Advanced high-profile techniques, such as solid surface arrays and display technologies, 16 have been boosting the biomarker discovery. Despite considerable progress and widespread application in autoantibody discovery, 17–19 the utility of high-throughput assays, particularly protein microarrays, remains restricted by the limited amount of miniaturised test sites. This constrains the coverage of the whole proteome and the detection sensitivity and specificity for individual protein. In addition, patient heterogeneity and varied abundance of targets can limit the progress in finding specific biomarkers in serum. Therefore, a comprehensive strategy with pre-enrichment of protein targets followed by subsequent whole-proteome screening for functional properties will facilitate the investigation of serum alterations in patients. 20 21 However, this approach necessitates extensive proof-of-concept studies and wide-spectrum cohort validation.
Here, we first identified PRMT5 as a novel autoantibody target in SSc based on the automated deep efficient peptide sequencing and quantification (DEEP SEQ) mass spectrometry (MS) platform 22 for the enriched antigen-antibody complex. We then validated the prevalence of antibodies against PRMT5 in the sera of patients with SSc and demonstrated the close correlations of anti-PRMT5 antibodies with the progression or regression trajectories of skin and lung disease in SSc. Induction of SSc-like skin and lung changes in mice via immunisation with recombinant PRMT5 protein indicates anti-PRMT5 as a potential contributing antibody in the pathogenesis of SSc. Anti-PRMT5 antibodies manifested as diagnostic and predictive marker for SSc. The utilisation of anti-PRMT5 antibodies has the potential to enhance precise disease monitoring and prognosis assessment in SSc.
Global autoantibody profiling in SSc via DEEP SEQ proteomics
To uncover elusive autoantibody surrogates for SSc, we developed a global antibody profiling strategy based on the automated DEEP SEQ MS platform 22 ( figures 1A and 2A ). In brief, we constructed an antigen pool using lysates from a variety of cell lines, including human umbilical vein endothelial cells, human dermal fibroblasts, Jurkat T cells and THP-1 monocytes, as sources of antigens for subsequent proteomics profiling. The corresponding cell types have been linked to the pathogenesis of SSc. 4 13 14 Antigen-antibody complexes were then enriched by coincubating global antibodies with the antigen pool, in conjunction with immunoprecipitation (IP) using protein A/G beads. Finally, we performed in-solution on-bead trypsin digest of the complexes and labelled peptide fragments, following subsequent quantitative proteomics analysis ( figure 2A ).
- Download figure
- Open in new tab
- Download powerpoint
Overview of experimental workflow and key discoveries. (A) Peripheral blood was obtained from the patients with SSc and healthy subjects (n=3/group), and then coincubated with an antigen pool, which was prepared from a diverse array of cell lysate of endothelial cells, fibroblasts, T cells and monocytes. Then the antigen-antibody complex was enriched with protein A/G and proceeded with on-bead digestion and labelling by means of isobaric tags for relative and absolute quantitation (iTRAQ). The quantitative proteomics of these pulldowns enabled us to illuminate the presence of over-represented putative antibody targets in patients. Applying the automated deep efficient peptide sequencing and quantification (DEEP SEQ) mass spectrometry (MS) platform, 37 our immunoprecipitation (IP)-MS analysis revealed putative autoantibodies against antigen pools, which require further verification to exclude non-specific binding. (B) Peripheral blood was obtained from 90 patients with systemic sclerosis (SSc) patients, 30 patients with systemic lupus erythematosus (SLE), 8 patients with Sjögren’s syndrome (SjS) and 84 healthy donors for antibody validation. Antibodies against PRMT1, PRMT5, HK1 and CD5L in the serum were determined by ELISA. Anti-PRMT5 was identified as a specific autoantibody for SSc. The expression of PRMT5 in SSc skin was evaluated by immunofluorescence (IF) staining. (C) The levels of anti-PRMT5 antibodies were compared between healthy subjects and patients with SSc and correlated with clinical phenotypes of patients with SSc. (D) Immunisation of mice with recombinant protein PRMT5 subcutaneously at an interval of 2 weeks for a total of four times. Skin and lung tissues were collected for pathological examination and bulk RNA sequencing (RNA-seq). Skin and lung fibrosis were found in PRMT5-immunised mice, which mimicked human SSc-like changes; thus, anti-PRMT5 antibody was identified as a potential contributing antibody for SSc.
Proteomics-based discovery of autoantibodies in SSc. (A) Schematic illustration of the proteomics-based approach employed to identify autoantibodies associated with SSc. The graphic was generated using BioRender.com and complies with BioRender’s Academic License Terms. (B) Heatmap diagram displaying differentially expressed antibodies in the sera of patients with SSc compared with healthy controls. Red values indicate upregulation, while green values indicate downregulation. (C) Identification of autoantibodies by proteomics in SSc. Volcano plots illustrate iTRAQ proteomics results for enriched and purified proteins bound to antibodies in serum from patients with SSc versus healthy donors. The x-axis represents relative protein levels (mean log2 iTRAQ ratios across three replicate experiments) in patients with SSc compared with healthy donors, while the y-axis displays log10 (p values). Significantly enriched and upregulated proteins (p≤0.05; iTRAQ ratio ≥1.5) are denoted by red dots, significantly enriched and downregulated proteins (p≤0.05; iTRAQ ratio ≤−1.5) by blue dots, and all others by grey dots. The dotted lines indicate a 1.5-fold ratio (x-axis) and a p-value of 0.05 (y-axis). (D) Protein-protein interaction networks of a subset of significantly enriched and upregulated antibodies in SSc, highlighting PRMT5 as the antibody target. (E) Heatmap diagram depicting representative differentially expressed antibodies in SSc. Red values indicate upregulated proteins, while blue values indicate downregulated proteins. PRMT5 as the target is highlighted. iTRAQ, isobaric tags for relative and absolute quantitation; HC, healthy control; MS, mass spectrometry; SSc, systemic sclerosis.
In this study, sera from patients with SSc (n=3) and healthy controls (HCs) (n=3) were subjected to the detection of global antibodies ( online supplemental table S1 ). By applying the DEEP SEQ proteomics platform, we identified 4798 proteins in total (≤1% false discovery rate (FDR) at peptide level). Among these, 238 enriched proteins were significantly upregulated in SSc in contrast to HCs in IP-MS analysis targeting putative autoantibodies against antigen pools ( figure 2B,C ). Not all of the 238 identified proteins serve as targets for autoantibodies; some may exhibit non-specific binding due to the limitations of quantitative proteomics techniques. Therefore, further verification is necessary to screen for potential autoantibody candidates within this group of proteins. To confirm the validity of antigen capture process, topoisomerase I (Topo I) was detected by western blotting following IP in a patient who was positive for anti-Topo I antibody (ATA), whereas undetectable in an ATA-negative patient ( online supplemental figure S1A,B ). In addition, protein-protein interaction networks showed the emergence of protein interaction ‘hot spots’ surrounding Topo I, one of the well-defined antibody targets and frequently associated with SSc 23 ( figure 2D ). Among Topo I-associated proteomes, PRMT1, PRMT5, HK-1 and CD5L were over-represented in patients with SSc than HC ( figure 2E ). Since literature results suggested PRMT1, PRMT5, HK-1 and CD5L with promising pathophysiological relevance, 24–28 we then focused our investigations on these four proteins as potential autoantibody targets.
Supplemental material
Validation of anti-prmt5 antibody as a specific autoantibody for ssc.
To confirm the discovery results, serum levels of antibodies against PRMT1, PRMT5, HK-1 and CD5L were then measured by ELISA in a primary validation cohort, including 90 patients with SSc, 30 patients with systemic lupus erythematosus (SLE), 8 patients with Sjögren’s syndrome (SjS) and 84 sex-matched and age-matched HC ( online supplemental table S2 , figure 1B ). Serial dilutions for serum were applied to determine the optimal condition for ELISA and calculate values of area under the curve (AUC) ( online supplemental figure S2A–D ). Serum levels of antibodies against PRMT5, measured as absorbance signals at 405 nm by ELISA, were significantly higher in patients with SSc, compared with HC or the patients with SLE and SjS (SSc vs HC p<0.001; SSc vs SLE p<0.001; SSc vs SjS p=0.003) ( figure 3A ). Consistently, when calculated by the values of AUC using serial dilutions, serum levels of anti-PRMT5 antibodies also demonstrated an increase in patients with SSc relative to HC, SLE or SjS ( online supplemental figure S3A ). On the contrary, the level of anti-CD5L antibodies showed a moderate increase in patients with SSc relative to HC (p=0.019) but was comparable with the patients of SLE or SjS. No significant difference was found in serum levels of antibodies against PRMT1 or HK1 across SSc, HC, SLE and SjS ( figure 3A , online supplemental figure S3B–D ). Next, to exclude technical false positivity, non-relevant anti-ZIKV envelope DIII virus antibodies 29 were tested as undetectable in both patients with SSc and HC ( online supplemental figure S4A–C ). Furthermore, using the 99th percentile as the upper limit of normal, anti-PRMT5 antibodies were present in 31.11% of patients with SSc (28/90) and absent in HC (0/84) ( figure 3B ), with sensitivity, specificity, positive predictive value and negative predictive value of 70.24%, 97.78%, 96.72% and 77.88%, respectively. The positivity of anti-PRMT5 antibodies in SSc was greater than anti-PRMT1, HK-1 and CD5L antibodies ( figure 3C–E , online supplemental figure S3E–G ). Interestingly, anti-PRMT5 antibodies also demonstrated the ability to differentiate SSc from the patients of SLE and SjS with AUC of 0.968 and 0.988, respectively ( figure 3D, E ). Following this, we proceeded to validate the levels of anti-PRMT5 antibodies in a recently recruited, independent group of patients with SSc. Consistently, we observed significantly elevated levels of anti-PRMT5 in the sera of these patients with SSc compared with the HC ( online supplemental table S3 , online supplemental figure S5A,B ). Furthermore, we validated the elevated levels of anti-PRMT5 antibodies in sera from these patients with SSc and HC determined using microarray ( online supplemental figure S5C,D ). Overall, these results indicate that the anti-PRMT5 antibody is a specific surrogate biomarker for SSc.
Validation of anti-PRMT5 antibody as a specific autoantibody for SSc. (A) Antibodies against PRMT1, PRMT5, HK-1 and CD5L, determined by ELISA, in serum of 90 patients with systemic sclerosis (SSc), 30 patients with systemic lupus erythematosus (SLE), 8 patients with Sjögren’s syndrome (SjS) and 84 healthy controls (HCs). Data of A are presented as median±IQR, each dot representing one sample. P values were determined by Kruskal-Wallis test with Dunn’s multiple post hoc tests. P values are indicated in the figures. (B) Bar graphs demonstrating the proportion of patients positive or negative for the antibodies against PRMT5, PRMT1, HK-1 and CD5L. The positivity of serum antibody levels was determined if the values were above the 99th percentile as the upper limit of healthy donors. P values were determined by Fisher’s exact test. (C–E) Illustrations of the receiver operating characteristic (ROC) curves, plotted based on the serum levels of antibodies against PRMT5, PRMT1, HK-1 and CD5L in patients with SSc, compared with HC (C), SLE (D) and SjS (E), respectively. The values of the area under the curve (AUC) represent as indicated.
Correlations of the serum levels of anti-PRMT5 antibodies with clinical features of SSc
To define the clinical implication of anti-PRMT5 antibodies, we compared the serum antibody levels in patients with SSc with an array of clinical phenotypes ( figure 1C ). Patients exhibiting progression in skin fibrosis, as determined by a 25% increase in the modified Rodnan skin score (mRSS) compared with the previous visit within a 12-month period, demonstrated elevated serum levels of anti-PRMT5 antibodies ( figure 4A ). With respect to the relation between anti-PRMT5 antibodies and skin or lung score trajectories of the patients with SSc, we investigated if the occurrence and dynamic changes of these antibodies may fluctuate in parallel with the skin and lung changes during the disease course. Follow-up investigations revealed an elevation in anti-PRMT5 antibodies among patients exhibiting progression in mRSS and a decline in those with mRSS regression ( figure 4B ). To assess the predictive value of baseline anti-PRMT5 antibody levels for skin fibrosis progression over a prospective 12-month period, we examined patients with SSc manifesting skin fibrosis progression, which was defined as an increase in mRSS ≥25% from baseline in the follow-up visit (12 months after baseline). The patients with SSc with mRSS progression displayed a numerical elevation in baseline levels (p=0.102) of anti-PRMT5 antibody compared with the patients with SSc without skin fibrosis progression ( figure 4C ). Anti-PRMT5 antibodies also demonstrated the ability to differentiate patients with SSc with mRSS progression from the patients with SSc without mRSS progression with an AUC of 0.792 ( figure 4D ). To test the potential of anti-PRMT5 antibody as a candidate disease indicator, we took a thorough follow-up for patients with SSc, assessing skin mRSS score and examining anti-PRMT5 levels every 3 months. Remarkably, we observed a parallel change between anti-PRMT5 levels and mRSS scores ( online supplemental figure S6A,B ).
Correlations between anti-PRMT5 antibody levels and the progression of skin and lung fibrosis in patients with systemic sclerosis (SSc). (A) Comparison of anti-PRMT5 antibody levels in patients with SSc, who showed progression or no progression defined by an increase (≥ 25%) in their total mRSS score in the past 12±2 months. (B) Paired comparison analysis of anti-PRMT5 antibody levels of patients with SSc with progression or regression in total mRSS, comparing between the baseline and the follow-up mRSS score with an interval of 12±2 months. (C) Comparison of anti-PRMT5 antibody levels in patients with SSc, with and without skin fibrosis progression, was evaluated prospectively. Skin fibrosis progression was defined as an increase in the modified Rodnan skin score (mRSS) of ≥25% from baseline in the follow-up visit (12 months after baseline). Comparison data are shown as bar graphs with individual values, where each dot represents one sample, and the median and quartiles are indicated. (D) Illustrations of the receiver operating characteristic (ROC) curves, plotted based on the serum levels of antibodies against PRMT5, comparing between patients with SSc with or without mRSS progression prospectively. The values of area under the curve (AUC) represent as indicated. (E) Comparison of anti-PRMT5 antibody levels in patients with SSc-interstitial lung disease (ILD), who showed progression or no progression in their high-resolution CT (HRCT), compared with the HRCT undertaken in the past 12±2 months. (F) Paired comparison analysis of anti-PRMT5 antibody levels of patients with SSc-ILD with progression or regression in lung HRCT, comparing the baseline and the follow-up lung HRCT within an interval of 12±2 months. (G) Comparison of anti-PRMT5 antibody levels in patients with SSc developing progressive fibrosing ILD (PF-ILD) or not in the preceding 24-month follow-up. Comparison data in A, C, E and G are shown as bar graphs with individual values, each dot representing one sample, with the median shown as a continuous line and the quartiles as discontinuous lines. Data in A, C, E and G were analysed by two-sided Mann-Whitney test. Data in B and F were analysed by Wilcoxon matched-pair signed rank test. The p values are indicated in the figures, and p<0.05 was considered statistically significant.
Likewise, patients having experienced progression in lung fibrosis, determined by an increased involvement of semiquantified areas in high-resolution CT (HRCT) in the past 12 months, demonstrated a significant elevation in anti-PRMT5 antibody levels ( figure 4E ). Similarly, the trends of anti-PRMT5 antibodies exhibited a parallel change in patients with SSc with HRCT progression or regression, as compared with the follow-up HRCT score ( figure 4F ). Patients with SSc who fulfilled the criteria of progressive fibrosing interstitial lung disease (ILD) (PF-ILD) in the subsequent 24-month follow-up demonstrated significantly increased basal levels of anti-PRMT5 antibodies compared with the patients with SSc without developing PF-ILD ( figure 4G ).
Furthermore, serum anti-PRMT5 antibodies correlated positively with the levels of acute phase reactants (APRs) like erythrocyte sedimentation rate (ESR) and C reactive protein (CRP), as well as IgG and tissue inhibitor of metal protease 1 (TIMP-1) in SSc ( figure 5A ). Furthermore, patients with SSc with elevated APR levels, defined as having at least one of the following, CRP ≥6 mg/L, ESR ≥28 mm per hour or platelet count ≥330×10⁹/L, showed higher serum levels of anti-PRMT5, compared with the patients with SSc without APR elevation ( figure 5B ). According to the criteria for active disease defined in focuSSced 30 study, we found that active patients with SSc also displayed a greater abundance of anti-PRMT5 antibody ( figure 5B ). Moreover, we found positive correlations between anti-PRMT5 antibody levels and concentrations of IL-6, tumour necrosis factor alpha, IL-10 and IL-8 in patients with SSc ( online supplemental figure S6C ). Together, our data may indicate a potential link between anti-PRMT5 antibody and the inflammatory status of patients with SSc.
Correlations between anti-PRMT5 antibody levels and inflammatory and autoimmune markers in patients with systemic sclerosis (SSc). (A) Correlations of the serum levels of anti-PRMT5 antibodies with erythrocyte sedimentation rate (ESR), C reactive protein (CRP), IgG and tissue inhibitor of matrix metalloproteinase (TIMP)-1 in SSc. (B) Comparison of anti-PRMT5 antibody levels between patients with SSc with elevated acute phase reactant (APR) levels and the individuals without, stratified as having at least one of the following or not: C reactive protein ≥6 mg/L, ESR ≥28 mm per hour or platelet count ≥330×10⁹/L (left). Comparison of anti-PRMT5 antibody levels between active patients with SSc and non-active patients with SSc, stratified according to the criteria for active disease defined in focuSSced 30 study (right). (C) Comparison of the positivity of anti-PRMT5 antibodies in patients with SSc with different clinical subsets, including diffuse cutaneous SSc (dcSSc) versus limited cutaneous SSc (lcSSc), positive versus negative for anti-topoisomerase I antibody (ATA), anticentromere antibody (ACA) and anti-RNA polymerase III antibody (ARA). (D) Comparison of percentages of interstitial lung disease (ILD) between patients with SSc double positive for both ATA and anti-PRMT5 antibodies (APA) or not (left). Comparison of percentages of dcSSc versus lcSSc between patients with SSc double positive for both ATA and APA or not (right). Data in A were analysed using non-parametric Spearman correlation analysis. Data in B were analysed by two-sided Mann-Whitney test. Contingency data in C and D were analysed using Fisher’s exact test. The p values are indicated in the figures, and p<0.05 was considered statistically significant.
Patients with SSc with diffuse cutaneous involvement (dcSSc) demonstrated a relatively higher positivity for anti-PRMT5 antibody compared with the patients with SSc with limited cutaneous involvement (lcSSc) but without statistical significance (dcSSc vs lcSSc: 56.34% vs 43.66%, p=0.179, figure 5C ). Additionally, no relevance was found between anti-PRMT5 antibody and other known SSc-specific antibodies, including ATA, ACA and ARA ( figure 5C ). Interestingly, 15 out of 90 (16.67%) patients were double positive for ATA and anti-PRMT5 antibodies. Notably, within this group, 13 out of 15 (86.67%) patients manifested evidence of ILD on HRCT, a higher proportion compared with the non-double positive patients (41/75, 54.67%), with statistical significance (p=0.023, figure 5D ). Interestingly, among 18 follow-up patients, all 3 individuals who tested positive for both ATA and anti-PRMT5 antibody experienced ILD progression and fulfilled the criteria of PF-ILD within the preceding 24-month follow-up. Additionally, patients with SSc who are double positive for ATA and anti-PRMT5 antibodies also exhibited a numerical predominance for the diffuse cutaneous subset (11/15, 73.33%) compared with the limited cutaneous subset (4/15, 26.67%) but without statistical significance ( figure 5D ). No correlations were found between serum levels of anti-PRMT5 antibodies with other clinical parameters in terms of age, sex, disease duration, therapeutic backgrounds, commodity diseases, current mRSS, the presence of digital ulcer (DU), pulmonary arterial hypertension (PAH), telangiectasia or the pattern of nailfold capillaroscopy (data not shown). The data suggest that anti-PRMT5 antibodies are more closely associated with the disease trajectory observed in the skin and lungs of patients with SSc, surpassing the correlation with their current level of involvement.
Furthermore, PRMT5 was more pronounced in fibroblasts and moderately increased in endothelial cells in the dermis of patients with SSc relative to HC ( online supplemental figure S7A–C ). As the apoptosis of endothelial cells contributes to the pathogenesis of SSc as one of the initial steps, 31 32 we also observed the significantly increased cell counts of apoptotic PRMT5-positive endothelial cells in the dermis of patients with SSc ( online supplemental figure S8 ), indicating the potential underlying mechanism that PRMT5 may be exposed from apoptotic endothelial cells, triggering autoimmune response subsequently. PRMT5 was observed to be expressed in CD3 + T cells and CD68 + macrophages of skin, however, without statistical difference between patients with SSc and HC (data not shown).
Induction of skin and lung fibrosis by immunisation with PRMT5
To elucidate the contribution of anti-PRMT5 antibodies to the development of SSc, we immunised mice with recombinant protein PRMT5 ( figure 6A ). Skin fibrosis and lung fibrosis were examined histopathologically 8 weeks after initiation of PRMT5 treatment. Treatment with PRMT5/complete Freund’s adjuvant (CFA), in contrast to the treatment with vehicle (Veh)/CFA, resulted in skin fibrosis with increased dermal thickness. In addition, there was no significant difference between the mice treated with PRMT5 and Topo I ( figure 6B,C ). Similarly, ILD was observed in PRMT5/CFA-treated mice, exhibiting extensive inflammatory infiltration and diffuse fibrosis, with remarkably increased Ashcroft score than Veh/CFA-treated control mice ( figure 6D,E ). In addition, the Ashcroft score was comparable between the mice treated with PRMT5 and Topo I ( figure 6D,E ). Immunofluorescence costaining showed an increased number of α-smooth muscle actin (αSMA + ) fibroblast activation protein (FAP + ) myofibroblasts in the skin and lungs of mice treated with PRMT5/CFA, compared with the control mice treated with Veh/CFA ( figure 6F–I ).
Induction of skin and lung fibrosis in mice immunised by recombinant protein PRMT5. (A) Immunisation with recombinant protein PRMT5 or DNA topoisomerase I (Topo I), along with complete Freund’s adjuvant (CFA) four times subcutaneously with an interval of 2 weeks. Skin and lung tissue samples were collected and followed by the pathological examination (n=6 independent biological samples per group). (B) Representative H&E and Masson’s trichrome staining of the skin shown at 200-fold magnification (scale bars=100 µm). (C) Quantification of dermal thickness, which are normalised to controls. (D) Representative H&E and Masson’s trichrome staining of the lungs shown at 200-fold magnification (scale bars=100 µm). (E) Ashcroft scores were assessed and normalised to controls. (F, H) Representative immunofluorescence staining for αSMA (green) and costaining with FAP (red) in the dermis (F) or lungs (H) of mice treated with PRMT5, or Topo I, along with CFA, at 400-fold magnification (F and H; scale bars=50 µm). (G, I) Numbers of αSMA-positive fibroblasts per high power field (HPF) in the skin (G) and lungs (I) are quantified. All data are presented as median±IQR, each dot representing one sample. P values were determined by Kruskal-Wallis test with Dunn’s multiple post hoc test (C, E, G and I). P values are indicated in the figures.
To further investigate the serological antibody response, we examined the induction of anti-PRMT5 or Topo I antibodies in sera of mice immunised with recombinant PRMT5 or Topo I, emulsified in CFA, through ELISA. Our findings revealed higher levels of anti-PRMT5 antibodies in sera of mice immunised with recombinant PRMT5, while higher levels of anti-Topo I antibodies in sera of mice immunised with recombinant Topo I ( online supplemental figure S9 ). Further multiplexed immunofluorescence staining was performed by labelling with antibodies against CD45, CD3, CD68 and CD20, which have been reported as markers for pronounced infiltrating immune cell types in SSc. 1 3 We found a remarkable increase in immune cell infiltration in the skin of PRMT5 immunised mice, including T cells, macrophages and B cells ( online supplemental figure S10A,B ). Immune infiltration dominant by T cells and macrophages was also found in the lungs of the mice immunised by PRMT5 plus CFA, compared with the Veh /CFA-treated control mice ( online supplemental figure S10C,D ).
To further explore the impact of anti-PRMT5 antibodies on mice and unravel underlying mechanism, we conducted RNA-seq analysis on skin and lung tissues obtained from the mice immunised with PRMT5/CFA or Veh/CFA. Applying a threshold of p<0.05 and |log2 fold change (FC)| ≥1, we identified a total of 4205 and 1169 differentially expressed genes (DEGs) in the skin and lungs, respectively (in the skin, 2681 upregulated DEGs and 1524 downregulated DEGs; in the lungs: 640 upregulated DEGs and 529 downregulated DEGs), comparing between PRMT5/CFA-treated mice versus Veh/CFA-treated control mice ( online supplemental figure S11A,B,E,F ). The gene sets encompass numerous genes previously implicated in the pathogenesis of SSc, such as Acta2 , Col1a1 , Col3a1 , Smad3 , Ctgf , Msr1 , Cd4 , Cd8a and Cd68 , supporting the potential of anti-PRMT5 in induction of SSc-like skin changes. Furthermore, the upregulated gene set identified in the lungs of PRMT5/CFA immunised mice included the overlapping DEGs observed in the skin, such as Smad3 , Msr1 and Cd8a , along with other fibrosis-associated genes like Cd163 , Shh , Edn1 and Fgf12 ( online supplemental figure S11E,F ).
To elucidate the functional associations of the identified gene signatures, we performed a gene enrichment analysis for Gene Ontology (GO). This analysis revealed the enrichment of genes involved in immune response and extracellular matrix organisation as key biological processes in the skin of PRMT5/CFA-immunised mice, suggestive of the activation of the immune system and fibrosis process that mirrors characteristics observed in patients with SSc ( online supplemental figure S11C ). Consistent with this notion, we observed the upregulation of the signalling pathways previously associated with SSc, 3 33 such as interleukin (IL)-6, IL-4, IL-17, IL-1β, toll-like receptor, vascular endothelial growth factor (VEGF) signalling, JAK-STAT signalling as well as pathways involved in wound healing and extracellular matrix organisation ( online supplemental figure S11C ). Further analysis using Reactome database demonstrated changes in VEGF or PDGF-mediated signalling, antigen presentation, extracellular matrix organisation, assembly of collagen fibrils as well as cell surface interaction at the vascular wall ( online supplemental figure S11D ).
In addition, systemic analysis using GO and Reactome databases demonstrated several terms relevant to SSc in the lungs of PRMT5/CFA-immunised mice, including ‘response to IL-13’, ‘wound healing involved in inflammatory response’, ‘response to hypoxia’, ‘antigen receptor-mediated signalling pathway’, ‘classical antibody-mediated complement activation’ and ‘immunoregulatory interactions between a lymphoid and a non-lymphoid cell’ ( online supplemental figure S11G,H ).
We then delved into signalling pathways governed by DEGs via further Ingenuity Pathway Analysis (IPA). Alongside the activation of numerous fundamental immune and inflammatory responses, as well as cytokine signalling in skin tissues from the mice immunised with PRMT5 ( online supplemental figure S12A ), our analysis pinpointed the upregulation of a cascade of fibrosis-associated signalling pathways integral to the pathogenesis process, 4 33 including STAT3, PDGF and G-proteincoupled receptor ( online supplemental figure S12 ). Signalling pathways associated with tissue and matrix regulation have been over-represented in the skin tissues of PRMT5 immunised mice, such as ‘wound healing signalling pathway’, ‘extracellular matrix organisation’ and ‘collagen biosynthesis and modifying enzymes’. Conversely, the suppression signal like ‘inhibition of matrix metalloproteases’ was significantly inhibited ( online supplemental figure S12A ). This comprehensive understanding is summarised through a graphical network encompassing canonical pathways, upstream regulators and biological functions ( online supplemental figure S12B ).
Therefore, immunisation with PRMT5 provokes SSc-mimicking inflammation and fibrosis in the skin and lungs in vivo. These findings may indicate PRMT5 as a potential contributing antibody target in SSc, likely participating in the processes of autoimmune response and myofibroblast activation.
Our study here is the first to identify PRMT5 as a novel autoantibody target of the autoimmune response in patients with SSc based on large-scale proteomics with automated DEEP SEQ. 22 Anti-PRMT5 antibodies are present in 31.11% of patients with SSc and absent in HCs. We found the dynamic changes of anti-PRMT5 antibodies in parallel with the progression or regression of skin fibrosis and ILD of SSc; monitoring the levels of anti-PRMT5 antibodies may therefore enable early detection and the initiation of early intervention for the patients with SSc with a higher risk of mRSS progression and development to PF-ILD during follow-up. Histopathological evaluations further revealed an overexpression of PRMT5, predominantly localised within fibroblasts and endothelial cells, among patients with SSc. Furthermore, mice immunised with PRMT5 showed marked tissue fibrosis coupled with immune cell infiltration within both dermal and pulmonary tissues, as well as serological antibody response, mirroring the pathognomonic changes typically associated with human SSc. RNA-seq demonstrated immunisation with PRMT5-induced multiple profibrotic and proinflammatory transcriptional networks in mice. Collectively, these data thus indicate the potential role of anti-PRMT5 as a contributing autoantibody in the development of SSc.
The first step of the current study is a screening for autoantigens with an exploratory strategy using high-throughput proteomics. Lysates of the cell lines were used as a source of putative autoantigens. The antibody-antigen complex was enriched via IP and analysed with large-scale proteomics based on automated DEEP SEQ MS as we established previously. 22 In contrast to the DNA sequencing platforms, the protein-level array poses challenges attributed to the wide dynamic span in protein expressions and vast diversity in post-translational modifications, coupled with the lack of an amplification strategy analogous to PCR, limiting genome-wide protein characterisation, particularly for signal transduction and other key regulatory factors that are often present in low abundance. To address this issue, we employed an established genome-scale proteome quantification by DEEP SEQ MS, which is based on simple detergent lysis and single-enzyme digest, extreme, orthogonal separation of peptides and true nanoflow liquid chromatography (LC)-MS/MS, significantly increasing the scale of proteome coverage. 22 In fact, omitting protein crosslinking may avoid the complexities associated with diverse protein conformations. Subsequent studies in patient cohort demonstrated that the autoantibodies against PRMT5 can be detected in SSc with high diagnostic performance. In this case, these results support the feasibility of this coordinated workflow combining thorough screen and functional validation for the identification of optimal diagnostic and therapeutic antibody candidates in general.
The presence of autoantibodies in patients does not mean that the autoantibodies can mediate the clinical manifestations. Therefore, the challenge is to clarify the role of autoreactivity in a clinical scenario and determine whether autoreactivity is crucial or merely incidental. The validity of anti-PRMT5 antibodies rather than anti-PRMT1, HK-1 or CD5L antibodies with higher specificity and sensitivity for SSc leads to the next in-depth investigation for anti-PRMT5 in SSc. Besides, anti-PRMT5 antibodies demonstrated promising predictive value for the progression of skin and lung disease in SSc. Therefore, the role of autoimmunity of anti-PRMT5 was then assessed in an immunisation model, which has been extensively used for model establishment, such as experimental allergic encephalomyelitis, 34 collagen-induced arthritis, 35 glucose-6-phosphate isomerase-induced arthritis 36 as well as Topo I immunised SSc. 23 Indeed, induction of tissue fibrosis and immune infiltration in mice immunised by recombinant protein PRMT5 illustrated that the antibody response to PRMT5 was likely to result in SSc-like manifestations. As anticipated, we observed several upregulated DEGs in he skin related to T cell, B cell and macrophage response, as well as fibroblast activation. Through GO and Reactome analysis, we revealed characteristic changes in both the skin and lungs of PRMT5-immunised mice, clearly distinguishing them from controls. These changes included several key functional categories: (1) we observed various biological processes in PRMT5-immunised mice related to autoimmune responses, with particular emphasis on processes linked to antigen presentation and immune cell activation; (2) we showed the increased production of profibrotic cytokines, including IL-4, IL-13, IL-6 and IL-1β; (3) we found multiple biological processes related to fibroblast activation and fibrotic tissue remodelling, including extracellular matrix organisation, biosynthesis and assembly; and (4) we observed the activity of several key profibrotic signalling, such as VEGF, PDGF, JAK-STAT and Toll-like receptor-mediated signalling. Further comprehensive analysis encompassing canonical pathways, upstream regulators and biological functions was also conducted by IPA. Thus, our unbiased RNA-seq analysis highlights immune response and tissue remodelling as a characteristic feature across the skin and lungs in the mice immunised with PRMT5, mirroring aspects of human SSc pathology.
The overexpression of PRMT5 in the fibroblasts and endothelial cells of SSc has provided the rationale that PRMT5 might be related to the fibroblast activation and endothelial dysfunction, which have been revealed as crucial in the pathogenesis of SSc. 2 4 PRMTs have been shown to play critical roles in disease through methylation of arginine residues on histone or non-histone proteins. Of note, circulating monomethyl arginine and asymmetrically dimethylated arginine can inhibit the function of nitric oxide (NO) synthase, which generates NO. Interestingly, attenuated NO bioavailability results in a milieu of inflammation and oxidative stress in SSc, leading to vasculopathy and subsequent fibrosis and reshaping of NO metabolism has been proven to be an effective treatment of SSc-associated vasculopathy, especially for DU and PAH. 37 38 PRMT5 was identified as a symmetrical dimethyltransferase ubiquitously expressed in the kidneys, skin, lungs and other tissues. 39 PRMT5 inhibitors have demonstrated efficiency in treating mouse models of acute graft-versus-host disease, as elucidated by prolonged survival and ameliorated disease severity, along with decreased T cell proliferation and cytokine production. 40 PRMT5 has been reported to regulate T cells through various pathways, including promoting retinoid-related orphan receptor (ROR)-γt activity and adjusting the Klf2-S1pr1 pathway. 41 42 Arginine methylation mediated by PRMTs has emerged as a critical mechanism implicated in fibrosis. 43–47 Notably, fibroblast-specific deletion of PRMT5 significantly reduced pressure overload-induced cardiac fibrosis. PRMT5 has been shown to regulate transforming growth factor beta (TGF-β)/Smad3-dependent fibrotic gene transcription, potentially through histone methylation crosstalk, and plays a critical role in cardiac fibrosis and dysfunction. 45 Similarly, the contribution of PRMT5 to fibrosis has been confirmed in an Adriamycin-induced cardiac fibrosis model. 46 These findings suggest that PRMT5 may serve as a critical mediator in regulating TGF-β-stimulated fibroblast activation and tissue fibrosis. Since protein methylation is a targetable modification and advanced drug development of PRMT5 inhibitors has been achieved, the therapeutic potential of targeting PRMT5 appears promising.
In sum, this study has identified anti-PRMT5 antibodies as a novel biomarker for SSc. The current data suggest the potential underlying mechanism driven by PRMT5 as a target of autoimmunity and consequently resulting fibrosis in SSc. However, the exact role of anti-PRMT5 in SSc needs further elucidation.
A detailed description of all materials and methods is provided in online supplemental material .
All human studies were approved by the ethical committee of the Medical Faculty of Fudan University. All patients and controls signed an informed consent form approved by the local institutional review board.
All animal experiments were carried out in strict accordance with international and local guidelines for animal care and use. Mice were maintained under pathogen-free conditions, with a standard diet, water ad libitum and 12 hour light/12 hour dark cycle. Mice were 6-week old at the start of experiments, and up to six mice were housed in one cage.
Statistical measures, including the number of samples, descriptive statistics (median and IQR) and significance, are reported in the figures and figure legends. P<0.05 was considered statistically significant.
Ethics statements
Patient consent for publication.
Not applicable.
Ethics approval
The protocol of mouse experiment was approved by the Fudan University, Shanghai, PRC (2021JS HSYY-052; 2022JS HSYY-054; 2023-HSYY-179JZS). This study involves human participants and was approved by Huashan Hospital Institutional Review Board. Participants gave informed consent to participate in the study before taking part.
Acknowledgments
We would particularly like to acknowledge my colleagues at the Division of Rheumatology of Huashan Hospital for their wonderful collaboration and patient support. We thank AbCode Co, Ltd, for the support of microarray assay.
- Gabrielli A ,
- Avvedimento EV ,
- Denton CP ,
- Chakraborty D ,
- Distler JHW
- Distler JHW ,
- Györfi A-H ,
- Ramanujam M , et al
- Patterson K , et al
- Pisetsky DS
- Nihtyanova SI ,
- Chen C , et al
- Wang X , et al
- McMahan ZH ,
- Kulkarni S ,
- Andrade F , et al
- Favoino E ,
- Cipriani P ,
- Liakouli V , et al
- Baroni SS ,
- Santillo M ,
- Bevilacqua F , et al
- Riemekasten G ,
- Pauling JD ,
- Salazar G ,
- Lu H , et al
- Clark KEN ,
- Attah M , et al
- Carlton LH ,
- McGregor R ,
- Moreland NJ
- Ho J , et al
- Sjöberg R ,
- Mattsson C ,
- Andersson E , et al
- Jaycox JR ,
- Vulsteke J-B ,
- Dubucquoi S , et al
- Storey AJ ,
- Hassen SI , et al
- Ficarro SB , et al
- Yoshizaki A ,
- Ogawa A , et al
- Sengupta S ,
- Kennemer A ,
- Patrick K , et al
- Edell C , et al
- Huang W , et al
- Iannaccone A ,
- Hollingsworth TJ ,
- Koirala D , et al
- Gaublomme J , et al
- Du L , et al
- Furst DE , et al
- Mostmans Y ,
- Giddelo C , et al
- Maehara T ,
- Perugino CA , et al
- Kerlero de Rosbo N ,
- Meehan GR ,
- Al Khabouri S , et al
- Krämer A , et al
- Girgis RE ,
- Gugnani MK ,
- Abrams J , et al
- Aubourg F , et al
- Fagerberg L ,
- Hallström BM , et al
- Snyder KJ ,
- Zitzer NC ,
- Gao Y , et al
- Kim H-Y , et al
- Zhou B , et al
- Zakrzewicz D ,
- Zakrzewicz A ,
- Didiasova M , et al
- Katanasaka Y ,
- Murata N , et al
- Yu S-Z , et al
- Liu F , et al
Supplementary materials
Supplementary data.
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
- Data supplement 1
- Data supplement 2
- Data supplement 3
- Data supplement 4
- Data supplement 5
- Data supplement 6
- Data supplement 7
- Data supplement 8
- Data supplement 9
- Data supplement 10
- Data supplement 11
- Data supplement 12
- Data supplement 13
Handling editor Josef S Smolen
ML, LW, XT, KW and XZ contributed equally.
Correction notice This article has been corrected since it published Online First. The legend for figure 3 and the funding statement have been corrected.
Contributors Overall content as the guarantors: MRL, TLY, FZ. Conceptualisation: ML, TY, FZ. Methodology: LBW, XLT, KW, QL, XYZ, TLY, FZ, XYZ. Investigation: MRL, LBW, XLT, KW, QL, XYZ, TLY, FZ. Visualisation: MRL, LBW, XLT, KW, XYZ. Funding acquisition: MRL, HJZ, TLY, FZ. Project administration: MRL, TLY, FZ. Supervision: MRL, TLY, FZ. Writing—original draft: MRL, FZ. Writing—review and editing: MRL, LBW, XLT, KW, XYZ, QL, LLH, WJY, HHY, XXZ, YX, WGW, YLW, XJC, LWL, JCW, HJZ, TLY, FZ.
Funding This study was also supported by the National Natural Science Foundation of China (NSFC) (82371818), Joint Sino-German research project from NSFC and Deutsche Forschungsgemeinschaft (German Research Foundation) (82161138022), NSFC (82030003), NSFC (32171432), National Key R&D Program of China (2022YFC3400202) and Chinese Academy of Medical Sciences Innovation Fund for Medical Sciences (2019-I2M-5-066).
Competing interests None declared.
Patient and public involvement Patients and/or the public were not involved in the design, or conduct, or reporting, or dissemination plans of this research.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.
Read the full text or download the PDF:
Thank you for visiting nature.com. You are using a browser version with limited support for CSS. To obtain the best experience, we recommend you use a more up to date browser (or turn off compatibility mode in Internet Explorer). In the meantime, to ensure continued support, we are displaying the site without styles and JavaScript.
- View all journals
- Explore content
- About the journal
- Publish with us
- Sign up for alerts
- Review Article
- Published: 09 March 2021
Antigen presentation in cancer: insights into tumour immunogenicity and immune evasion
- Suchit Jhunjhunwala ORCID: orcid.org/0000-0003-3484-0588 1 ,
- Christian Hammer ORCID: orcid.org/0000-0003-4548-7548 1 &
- Lélia Delamarre ORCID: orcid.org/0000-0001-9122-5181 1
Nature Reviews Cancer volume 21 , pages 298–312 ( 2021 ) Cite this article
46k Accesses
525 Citations
115 Altmetric
Metrics details
- Cancer immunotherapy
- Cancer microenvironment
- Immunosurveillance
Immune checkpoint blockade, which blocks inhibitory signals of T cell activation, has shown tremendous success in treating cancer, although success still remains limited to a fraction of patients. To date, clinically effective CD8 + T cell responses appear to target predominantly antigens derived from tumour-specific mutations that accumulate in cancer, also called neoantigens. Tumour antigens are displayed on the surface of cells by class I human leukocyte antigens (HLA-I). To elicit an effective antitumour response, antigen presentation has to be successful at two distinct events: first, cancer antigens have to be taken up by dendritic cells (DCs) and cross-presented for CD8 + T cell priming. Second, the antigens have to be directly presented by the tumour for recognition by primed CD8 + T cells and killing. Tumours exploit multiple escape mechanisms to evade immune recognition at both of these steps. Here, we review the tumour-derived factors modulating DC function, and we summarize evidence of immune evasion by means of quantitative modulation or qualitative alteration of the antigen repertoire presented on tumours. These mechanisms include modulation of antigen expression, HLA-I surface levels, alterations in the antigen processing and presentation machinery in tumour cells. Lastly, as complete abrogation of antigen presentation can lead to natural killer (NK) cell-mediated tumour killing, we also discuss how tumours can harbour antigen presentation defects and still evade NK cell recognition.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout

Similar content being viewed by others

Antigen presentation in cancer — mechanisms and clinical implications for immunotherapy

MHC-II neoantigens shape tumour immunity and response to immunotherapy

Dendritic cells as orchestrators of anticancer immunity and immunotherapy
Data availability.
The data that support the findings of this study are available as Supplementary Figures and in cBioportal: https://www.cbioportal.org/ .
Waldman, A. D., Fritz, J. M. & Lenardo, M. J. A guide to cancer immunotherapy: from T cell basic science to clinical practice. Nat. Rev. Immunol. 20 , 651–668 (2020).
Article CAS PubMed PubMed Central Google Scholar
Sharpe, A. H. & Pauken, K. E. The diverse functions of the PD1 inhibitory pathway. Nat. Rev. Immunol. 18 , 153–167 (2018).
Article CAS PubMed Google Scholar
Chen, D. S. & Mellman, I. Oncology meets immunology: the cancer-immunity cycle. Immunity 39 , 1–10 (2013).
Article PubMed CAS Google Scholar
Chapuis, A. G. et al. T cell receptor gene therapy targeting WT1 prevents acute myeloid leukemia relapse post-transplant. Nat. Med. 25 , 1064–1072 (2019).
Lowe, K. L. et al. Novel TCR-based biologics: mobilising T cells to warm ‘cold’ tumours. Cancer Treat. Rev. 77 , 35–43 (2019).
Sahin, U. et al. An RNA vaccine drives immunity in checkpoint-inhibitor-treated melanoma. Nature 585 , 107–112 (2020).
Choi, Y. J. et al. A phase II, prospective, randomized, multicenter, open-label study of GX-188E, an HPV DNA vaccine, in patients with cervical intraepithelial neoplasia 3. Clin. Cancer Res. 26 , 1616–1623 (2020).
Glisson, B. et al. 1136O - Nivolumab and ISA 101 HPV vaccine in incurable HPV-16 + cancer. Ann. Oncol. 28 , v403–v404 (2017).
Article Google Scholar
Youn, J. W. et al. Pembrolizumab plus GX-188E therapeutic DNA vaccine in patients with HPV-16-positive or HPV-18-positive advanced cervical cancer: interim results of a single-arm, phase 2 trial. Lancet Oncol. 21 , 1653–1660 (2020).
Lu, Y.-C. et al. Efficient identification of mutated cancer antigens recognized by T cells associated with durable tumor regressions. Clin. Cancer Res. 20 , 3401310 (2014).
Rizvi, N. A. et al. Mutational landscape determines sensitivity to PD-1 blockade in non–small cell lung cancer. Science 348 , 124–128 (2015).
Rosenberg, J. E. et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: a single-arm, multicentre, phase 2 trial. Lancet 387 , 1909–1920 (2016).
Snyder, A. et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N. Engl. J. Med. 371 , 2189–2199 (2014). The first study to show that tumour mutation burden is associated with response to ICI in patients with melanoma .
Article PubMed PubMed Central CAS Google Scholar
Tran, E. et al. T-cell transfer therapy targeting mutant KRAS in cancer. N. Engl. J. Med. 375 , 2255–2262 (2016). Infusion of T cells against the mutation KRAS G12D presented by HLA-C802 leads to objective regression of metastases in a patient with colorectal cancer .
Zacharakis, N. et al. Immune recognition of somatic mutations leading to complete durable regression in metastatic breast cancer. Nat. Med. 24 , 724–730 (2018).
Carreno, B. M. et al. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science 348 , 803–808 (2015).
Hilf, N. et al. Actively personalized vaccination trial for newly diagnosed glioblastoma. Nature 565 , 240–245 (2019).
Keskin, D. B. et al. Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature 565 , 234–239 (2019).
Ott, P. A. et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 547 , 217–221 (2017).
Sahin, U. et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 547 , 222–226 (2017).
Wculek, S. K. et al. Dendritic cells in cancer immunology and immunotherapy. Nat. Rev. Immunol. 20 , 7–24 (2020).
Roberts, E. W. et al. Critical role for CD103 + /CD141 + dendritic cells bearing CCR7 for tumor antigen trafficking and priming of T cell immunity in melanoma. Cancer Cell 30 , 324–336 (2016).
Böttcher, J. P. & Reis e Sousa, C. The role of type 1 conventional dendritic cells in cancer immunity. Trends Cancer 4 , 784–792 (2018).
Hildner, K. et al. Batf3 deficiency reveals a critical role for CD8α + dendritic cells in cytotoxic T cell immunity. Science 322 , 1097–1100 (2008).
Salmon, H. et al. Expansion and activation of CD103 + dendritic cell progenitors at the tumor site enhances tumor responses to therapeutic PD-L1 and BRAF inhibition. Immunity 44 , 924–938 (2016).
Sánchez-Paulete, A. R. et al. Cancer immunotherapy with immunomodulatory anti-CD137 and anti-PD-1 monoclonal antibodies requires BATF3-dependent dendritic cells. Cancer Discov. 6 , 71–79 (2016).
Broz, M. L. et al. Dissecting the tumor myeloid compartment reveals rare activating antigen-presenting cells critical for T cell immunity. Cancer Cell 26 , 638–652 (2014).
Mayoux, M. et al. Dendritic cells dictate responses to PD-L1 blockade cancer immunotherapy. Sci. Transl Med. 12 , eaav7431 (2020).
Enamorado, M. et al. Enhanced anti-tumour immunity requires the interplay between resident and circulating memory CD8 + T cells. Nat. Commun. 8 , 16073 (2017).
Spranger, S., Dai, D., Horton, B. & Gajewski, T. F. Tumor-residing Batf3 dendritic cells are required for effector T cell trafficking and adoptive T cell therapy. Cancer Cell 31 , 711–723.e4 (2017). Intratumoural DCs are essential for the trafficking of effector T cells to the tumour through the secretion of the chemokines CXCL9 and CXCL10 .
Garris, C. S. et al. Successful anti-PD-1 cancer immunotherapy requires T cell-dendritic cell crosstalk involving the cytokines IFN-γ and IL-12. Immunity 49 , 1148–1161.e7 (2018).
Menares, E. et al. Tissue-resident memory CD8 + T cells amplify anti-tumor immunity by triggering antigen spreading through dendritic cells. Nat. Commun. 10 , 4401 (2019).
Ferris, S. T. et al. cDC1 prime and are licensed by CD4 + T cells to induce anti-tumour immunity. Nature 584 , 624–629 (2020).
Binnewies, M. et al. Unleashing type-2 dendritic cells to drive protective antitumor CD4 + T cell immunity. Cell 177 , 556–571.e16 (2019).
Spranger, S., Bao, R. & Gajewski, T. F. Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature 523 , 231–235 (2015).
de Galarreta, M. R. et al. β-Catenin activation promotes immune escape and resistance to anti-PD-1 therapy in hepatocellular carcinoma. Cancer Discov. 9 , 1124–1141 (2019).
Böttcher, J. P. et al. NK cells stimulate recruitment of cDC1 into the tumor microenvironment promoting cancer immune control. Cell 172 , 1022–1037.e14 (2018).
Lavin, Y. et al. Innate immune landscape in early lung adenocarcinoma by paired single-cell analyses. Cell 169 , 750–765.e17 (2017).
Zelenay, S. et al. Cyclooxygenase-dependent tumor growth through evasion of immunity. Cell 162 , 1257–1270 (2015).
Bonavita, E. et al. Antagonistic inflammatory phenotypes dictate tumor fate and response to immune checkpoint blockade. Immunity 53 , 1215–1229.e8 (2020).
Barry, K. C. et al. A natural killer–dendritic cell axis defines checkpoint therapy–responsive tumor microenvironments. Nat. Med. 24 , 1178–1191 (2018).
Galluzzi, L., Buqué, A., Kepp, O., Zitvogel, L. & Kroemer, G. Immunogenic cell death in cancer and infectious disease. Nat. Rev. Immunol. 17 , 97–111 (2017).
Sancho, D. et al. Identification of a dendritic cell receptor that couples sensing of necrosis to immunity. Nature 458 , 899–903 (2009).
Vacchelli, E. et al. Chemotherapy-induced antitumor immunity requires formyl peptide receptor 1. Science 350 , 972–978 (2015).
Yatim, N. et al. RIPK1 and NF-κB signaling in dying cells determines cross-priming of CD8 + T cells. Science 350 , 328–334 (2015).
Hangai, S. et al. PGE2 induced in and released by dying cells functions as an inhibitory DAMP. Proc. Natl Acad. Sci. USA 113 , 3844–3849 (2016).
Gabrilovich, D. I. et al. Production of vascular endothelial growth factor by human tumors inhibits the functional maturation of dendritic cells. Nat. Med. 2 , 1096–1103 (1996).
Ohm, J. E. & Carbone, D. P. VEGF as a mediator of tumor-associated immunodeficiency. Immunol. Res. 23 , 263–272 (2001).
Papaspyridonos, M. et al. Id1 suppresses anti-tumour immune responses and promotes tumour progression by impairing myeloid cell maturation. Nat. Commun. 6 , 6840 (2015).
Park, S.-J. et al. IL-6 regulates in vivo dendritic cell differentiation through STAT3 activation. J. Immunol. 173 , 3844–3854 (2004).
Yang, A. S. & Lattime, E. C. Tumor-induced interleukin 10 suppresses the ability of splenic dendritic cells to stimulate CD4 and CD8 T-cell responses. Cancer Res. 63 , 2150–2157 (2003).
CAS PubMed Google Scholar
Maier, B. et al. A conserved dendritic-cell regulatory program limits antitumour immunity. Nature 580 , 257–262 (2020). mregDCs, defined by expression of immunoregulatory and maturation genes, are a novel DC subset induced by tumour-antigen uptake. mregDCs reduce antitumour response .
Herber, D. L. et al. Lipid accumulation and dendritic cell dysfunction in cancer. Nat. Med. 16 , 880–886 (2010).
Cao, W. et al. Oxidized lipids block antigen cross-presentation by dendritic cells in cancer. J. Immunol. 192 , 2920–2931 (2014).
Article CAS Google Scholar
Cubillos-Ruiz, J. R. et al. ER stress sensor XBP1 controls anti-tumor immunity by disrupting dendritic cell homeostasis. Cell 161 , 1527–1538 (2015).
Veglia, F. et al. Lipid bodies containing oxidatively truncated lipids block antigen cross-presentation by dendritic cells in cancer. Nat. Commun. 8 , 2122 (2017).
Aznar, M. A. et al. Immunotherapeutic effects of intratumoral nanoplexed poly I:C. J. Immunother. Cancer 7 , 116 (2019).
Article PubMed PubMed Central Google Scholar
Corrales, L. et al. Direct activation of STING in the tumor microenvironment leads to potent and systemic tumor regression and immunity. Cell Rep. 11 , 1018–1030 (2015).
Ramanjulu, J. M. et al. Design of amidobenzimidazole STING receptor agonists with systemic activity. Nature 564 , 439–443 (2018).
Osada, T. et al. The effect of anti-VEGF therapy on immature myeloid cell and dendritic cells in cancer patients. Cancer Immunol. Immunother. 57 , 1115–1124 (2008).
Hammerich, L. et al. Systemic clinical tumor regressions and potentiation of PD1 blockade with in situ vaccination. Nat. Med. 25 , 814–824 (2019).
Lai, J. et al. Adoptive cellular therapy with T cells expressing the dendritic cell growth factor Flt3L drives epitope spreading and antitumor immunity. Nat. Immunol. 21 , 914–926 (2020).
Williford, J.-M. et al. Recruitment of CD103 + dendritic cells via tumor-targeted chemokine delivery enhances efficacy of checkpoint inhibitor immunotherapy. Sci. Adv. 5 , eaay1357 (2019).
Rock, K. L., Reits, E. & Neefjes, J. Present yourself! By MHC class I and MHC class II molecules. Trends Immunol. 37 , 724–737 (2016).
Wong, G. H., Clark-Lewis, I., McKimm-Breschkin, L., Harris, A. W. & Schrader, J. W. Interferon-gamma induces enhanced expression of Ia and H-2 antigens on B lymphoid, macrophage, and myeloid cell lines. J. Immunol. 131 , 788–793 (1983).
Klar, D. & Hämmerling, G. J. Induction of assembly of MHC class I heavy chains with beta 2microglobulin by interferon-gamma. EMBO J. 8 , 475–481 (1989).
Restifo, N. P. et al. Identification of human cancers deficient in antigen processing. J. Exp. Med. 177 , 265–272 (1993).
Shankaran, V. et al. IFNγ and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature 410 , 1107–1111 (2001).
Schreiber, R. D., Old, L. J. & Smyth, M. J. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science 331 , 1565–1570 (2011). Overview of cancer immunoediting, refinement of an initial review from 2002 .
Matsushita, H. et al. Cancer exome analysis reveals a T-cell-dependent mechanism of cancer immunoediting. Nature 482 , 400–404 (2012).
Maeurer, M. J. et al. Tumor escape from immune recognition: lethal recurrent melanoma in a patient associated with downregulation of the peptide transporter protein TAP-1 and loss of expression of the immunodominant MART-1/melan-A antigen. J. Clin. Invest. 98 , 1633–1641 (1996).
Russo, V. et al. Expression of the mage gene family in primary and metastatic human breast cancer: implications for tumor antigen-specific immunotherapy. Int. J. Cancer 64 , 216–221 (1995).
Rosenthal, R. et al. Neoantigen-directed immune escape in lung cancer evolution. Nature 567 , 479–485 (2019). TRACERx consortium study, analysing a clinical NSCLC cohort for LOH at HLA-I, and several modes of disruption to antigen presentation, and the association of these features with immune infiltration and disease-free survival .
Anagnostou, V. et al. Evolution of neoantigen landscape during immune checkpoint blockade in non-small cell lung cancer. Cancer Discov. 7 , 264–276 (2017).
Bassani-Sternberg, M., Pletscher-Frankild, S., Jensen, L. J. & Mann, M. Mass spectrometry of human leukocyte antigen class I peptidomes reveals strong effects of protein abundance and turnover on antigen presentation. Mol. Cell. Proteom. 14 , 658–673 (2015).
Jaeger, A. M. et al. Rebalancing protein homeostasis enhances tumor antigen presentation. Clin. Cancer Res. 25 , 6392–6405 (2019).
Rooney, M. S., Shukla, S. A., Wu, C. J., Getz, G. & Hacohen, N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 160 , 48–61 (2015). One of the first studies to use a large data set like TCGA to show that HLA and B2M mutations are associated with cytolytic activity, suggesting immune selection pressure .
Shukla, S. A. et al. Comprehensive analysis of cancer-associated somatic mutations in class I HLA genes. Nat. Biotechnol. 33 , 1152–1158 (2015). Extensive analysis of HLA mutations in TCGA data, showing prevalence of HLA-I mutations in several cancer types, and the landscape of mutations found in HLA-I genes .
Challa-Malladi, M. et al. Combined genetic inactivation of β2-microglobulin and CD58 reveals frequent escape from immune recognition in diffuse large B cell lymphoma. Cancer Cell 20 , 728–740 (2011).
Charette, M. de & Houot, R. Hide or defend, the two strategies of lymphoma immune evasion: potential implications for immunotherapy. Haematologica 103 , 1256–1268 (2018).
Middha, S. et al. Majority of B2M-mutant and -deficient colorectal carcinomas achieve clinical benefit from immune checkpoint inhibitor therapy and are microsatellite instability-high. JCO Precis. Oncol. 3 , PO.18.00321 (2019).
PubMed Central Google Scholar
Castro, A. et al. Elevated neoantigen levels in tumors with somatic mutations in the HLA-A, HLA-B, HLA-C and B2M genes. BMC Med. Genomics 12 , 107 (2019).
Sade-Feldman, M. et al. Resistance to checkpoint blockade therapy through inactivation of antigen presentation. Nat. Commun. 8 , 1136 (2017).
Allen, E. M. V. et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 350 , 207–211 (2015).
Zaretsky, J. M. et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N. Engl. J. Med. 375 , 819–829 (2016).
McGranahan, N. et al. Allele-specific HLA loss and immune escape in lung cancer evolution. Cell 171 , 1259–1271.e11 (2017).
Chen, Y. et al. Loss of heterozygosity at the human leukocyte antigen locus in thymic epithelial tumors. Thorac. Cancer 6 , 749–753 (2015).
Montesion, M. et al. Somatic HLA class I loss is a widespread mechanism of immune evasion which refines the use of tumor mutational burden as a biomarker of checkpoint inhibitor response. Cancer Discov. 11 , 282–292 (2021). Study examining a large pan-cancer clinical cohort for HLA loss and its association with clinical end points .
Lopez-Yrigoyen, M., Cassetta, L. & Pollard, J. W. Macrophage targeting in cancer. Ann. N. Y. Acad. Sci. https://doi.org/10.1111/nyas.14377 (2020).
Article PubMed Google Scholar
Guedan, S., Ruella, M. & June, C. H. Emerging cellular therapies for cancer. Annu. Rev. Immunol. 37 , 145–171 (2019).
Maude, S. L. et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 371 , 1507–1517 (2014).
Kantarjian, H. et al. Blinatumomab versus chemotherapy for advanced acute lymphoblastic leukemia. N. Engl. J. Med. 376 , 836–847 (2017).
Crowther, M. D. et al. Genome-wide CRISPR–Cas9 screening reveals ubiquitous T cell cancer targeting via the monomorphic MHC class I-related protein MR1. Nat. Immunol. 21 , 178–185 (2020).
Nangalia, J. et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N. Engl. J. Med. 369 , 2391–2405 (2013).
Arshad, N. & Cresswell, P. Tumor-associated calreticulin variants functionally compromise the peptide loading complex and impair its recruitment of MHC-I. J. Biol. Chem. 293 , 9555–9569 (2018).
Bozkus, C. C. et al. Immune checkpoint blockade enhances shared neoantigen-induced T-cell immunity directed against mutated calreticulin in myeloproliferative neoplasms. Cancer Discov. 9 , 1192–1207 (2019).
Article CAS PubMed Central Google Scholar
Arantes, A. Q. et al. Decreased activity of NK cells in myeloproliferative neoplasms. Blood 126 , 1637–1637 (2015).
Schönberg, K. et al. JAK inhibition impairs NK cell function in myeloproliferative neoplasms. Cancer Res. 75 , 2187–2199 (2015).
Dunn, G. P., Koebel, C. M. & Schreiber, R. D. Interferons, immunity and cancer immunoediting. Nat. Rev. Immunol. 6 , 836–848 (2006).
Manguso, R. T. et al. In vivo CRISPR screening identifies Ptpn2 as a cancer immunotherapy target. Nature 547 , 413–418 (2017).
Patel, S. J. et al. Identification of essential genes for cancer immunotherapy. Nature 548 , 537–542 (2017).
Gao, J. et al. Loss of IFN-γ pathway genes in tumor cells as a mechanism of resistance to anti-CTLA-4 therapy. Cell 167 , 397–404.e9 (2016).
Albacker, L. A. et al. Loss of function JAK1 mutations occur at high frequency in cancers with microsatellite instability and are suggestive of immune evasion. PLoS ONE 12 , e0176181 (2017).
Shin, D. S. et al. Primary resistance to PD-1 blockade mediated by JAK1/2 mutations. Cancer Discov. 7 , 188–201 (2017).
Mojic, M., Takeda, K. & Hayakawa, Y. The dark side of IFN-γ: its role in promoting cancer immunoevasion. Int. J. Mol. Sci. 19 , 89 (2017).
Article PubMed Central CAS Google Scholar
Benci, J. L. et al. Tumor interferon signaling regulates a multigenic resistance program to immune checkpoint blockade. Cell 167 , 1540–1554.e12 (2016).
Romero, J. M. et al. Coordinated downregulation of the antigen presentation machinery and HLA class I/β2-microglobulin complex is responsible for HLA-ABC loss in bladder cancer. Int. J. Cancer 113 , 605–610 (2005).
Meissner, T. B. et al. NLR family member NLRC5 is a transcriptional regulator of MHC class I genes. Proc. Natl Acad. Sci. USA 107 , 13794–13799 (2010).
Meissner, T. B., Li, A. & Kobayashi, K. S. NLRC5: a newly discovered MHC class I transactivator (CITA). Microbes Infect. 14 , 477–484 (2012).
Yoshihama, S. et al. NLRC5/MHC class I transactivator is a target for immune evasion in cancer. Proc. Natl Acad. Sci. USA 113 , 5999–6004 (2016).
Chew, G.-L. et al. DUX4 suppresses MHC class I to promote cancer immune evasion and resistance to checkpoint blockade. Dev. Cell 50 , 658–671.e7 (2019).
Ye, Q. et al. Hypermethylation of HLA class I gene is associated with HLA class I down-regulation in human gastric cancer. Tissue Antigens 75 , 30–39 (2010).
Qifeng, S., Bo, C., Xingtao, J., Chuanliang, P. & Xiaogang, Z. Methylation of the promoter of human leukocyte antigen class I in human esophageal squamous cell carcinoma and its histopathological characteristics. J. Thorac. Cardiovasc. Surg. 141 , 808–814 (2011).
Burr, M. L. et al. An evolutionarily conserved function of polycomb silences the MHC class I antigen presentation pathway and enables immune evasion in cancer. Cancer Cell 36 , 385–401.e8 (2019).
Ennishi, D. et al. Molecular and genetic characterization of MHC deficiency identifies EZH2 as therapeutic target for enhancing immune recognition. Cancer Discov. 9 , 546–563 (2019).
Yamamoto, K. et al. Autophagy promotes immune evasion of pancreatic cancer by degrading MHC-I. Nature 581 , 100–105 (2020).
Zebertavage, L. K., Alice, A., Crittenden, M. R. & Gough, M. J. Transcriptional upregulation of NLRC5 by radiation drives STING- and interferon-independent MHC-I expression on cancer cells and T cell cytotoxicity. Sci. Rep. 10 , 7376 (2020).
Chang, C.-C. et al. Multiple structural and epigenetic defects in the human leukocyte antigen class I antigen presentation pathway in a recurrent metastatic melanoma following immunotherapy. J. Biol. Chem. 290 , 26562–26575 (2015).
Aki, M. et al. Interferon-gamma induces different subunit organizations and functional diversity of proteasomes. J. Biochem. 115 , 257–269 (1994).
Driscoll, J., Brown, M. G., Finley, D. & Monaco, J. J. MHC-linked LMP gene products specifically alter peptidase activities of the proteasome. Nature 365 , 262–264 (1993).
Gaczynska, M., Rock, K. L. & Goldberg, A. L. Gamma-interferon and expression of MHC genes regulate peptide hydrolysis by proteasomes. Nature 365 , 264–267 (1993).
Kalaora, S. et al. Immunoproteasome expression is associated with better prognosis and response to checkpoint therapies in melanoma. Nat. Commun. 11 , 896 (2020).
Chapatte, L. et al. Processing of tumor-associated antigen by the proteasomes of dendritic cells controls in vivo T-cell responses. Cancer Res. 66 , 5461–5468 (2006).
Chapiro, J. et al. Destructive cleavage of antigenic peptides either by the immunoproteasome or by the standard proteasome results in differential antigen presentation. J. Immunol. 176 , 1053–1061 (2006).
Morel, S. et al. Processing of some antigens by the standard proteasome but not by the immunoproteasome results in poor presentation by dendritic cells. Immunity 12 , 107–117 (2000).
Evnouchidou, I. & van Endert, P. Peptide trimming by endoplasmic reticulum aminopeptidases: role of MHC class I binding and ERAP dimerization. Hum. Immunol. 80 , 290–295 (2019).
Chen, L. et al. Critical role of endoplasmic reticulum aminopeptidase 1 in determining the length and sequence of peptides bound and presented by HLA-B27. Arthritis Rheumatol. 66 , 284–294 (2014).
Nagarajan, N. A. et al. ERAAP shapes the peptidome associated with classical and nonclassical MHC class I molecules. J. Immunol. 197 , 1035–1043 (2016).
Reeves, E. & James, E. The role of polymorphic ERAP1 in autoinflammatory disease. Biosci. Rep. 38 , BSR20171503 (2018).
López de Castro, J. A. How ERAP1 and ERAP2 shape the peptidomes of disease-associated MHC-I proteins. Front. Immunol. 9 , 2463 (2018).
Stratikos, E., Stamogiannos, A., Zervoudi, E. & Fruci, D. A role for naturally occurring alleles of endoplasmic reticulum aminopeptidases in tumor immunity and cancer pre-disposition. Front. Oncol. 4 , 363 (2014).
Compagnone, M., Cifaldi, L. & Fruci, D. Regulation of ERAP1 and ERAP2 genes and their disfunction in human cancer. Hum. Immunol. 80 , 318–324 (2019).
Fruci, D. et al. Expression of endoplasmic reticulum aminopeptidases in EBV-B cell lines from healthy donors and in leukemia/lymphoma, carcinoma, and melanoma cell lines. J. Immunol. 176 , 4869–4879 (2006).
Textor, A. et al. Preventing tumor escape by targeting a post-proteasomal trimming independent epitope. J. Exp. Med. 213 , 2333–2348 (2016).
Lim, Y. W. et al. Germline genetic polymorphisms influence tumor gene expression and immune cell infiltration. Proc. Natl Acad. Sci. USA 115 , E11701–E11710 (2018).
van Deutekom, H. W. M. & Keşmir, C. Zooming into the binding groove of HLA molecules: which positions and which substitutions change peptide binding most? Immunogenetics 67 , 425–436 (2015).
Illing, P. T. et al. HLA-B57 micropolymorphism defines the sequence and conformational breadth of the immunopeptidome. Nat. Commun. 9 , 4693 (2018).
Garrido, G. et al. Tumor-targeted silencing of the peptide transporter TAP induces potent antitumor immunity. Nat. Commun. 10 , 3773 (2019).
Hammer, G. E., Gonzalez, F., James, E., Nolla, H. & Shastri, N. In the absence of aminopeptidase ERAAP, MHC class I molecules present many unstable and highly immunogenic peptides. Nat. Immunol. 8 , 101–108 (2007).
James, E., Bailey, I., Sugiyarto, G. & Elliott, T. Induction of protective antitumor immunity through attenuation of ERAAP function. J. Immunol. 190 , 5839–5846 (2013).
Keller, M. et al. The proteasome immunosubunits, PA28 and ER-aminopeptidase 1 protect melanoma cells from efficient MART-126-35-specific T-cell recognition. Eur. J. Immunol. 45 , 3257–3268 (2015).
Li, L. et al. Cross-dressed CD8α + /CD103 + dendritic cells prime CD8 + T cells following vaccination. Proc. Natl Acad. Sci. USA 109 , 12716–12721 (2012).
Das Mohapatra, A. et al. Cross-dressing of CD8α + dendritic cells with antigens from live mouse tumor cells is a major mechanism of cross-priming. Cancer Immunol. Res. 8 , 1287–1299 (2020).
Shimasaki, N., Jain, A. & Campana, D. NK cells for cancer immunotherapy. Nat. Rev. Drug Discov. 19 , 200–218 (2020).
Bald, T., Krummel, M. F., Smyth, M. J. & Barry, K. C. The NK cell-cancer cycle: advances and new challenges in NK cell-based immunotherapies. Nat. Immunol. 21 , 835–847 (2020).
Capuano, C. et al. Memory NK cell features exploitable in anticancer immunotherapy. J. Immunol. Res. 2019 , 8795673 (2019).
Guillerey, C., Huntington, N. D. & Smyth, M. J. Targeting natural killer cells in cancer immunotherapy. Nat. Immunol. 17 , 1025–1036 (2016).
Sivori, S. et al. Human NK cells: surface receptors, inhibitory checkpoints, and translational applications. Cell. Mol. Immunol. 16 , 430–441 (2019).
Dogra, P. et al. Tissue determinants of human NK cell development, function, and residence. Cell 180 , 749–763.e13 (2020).
Cózar, B. et al. Tumor-infiltrating natural killer cells. Cancer Discov. 11 , 34–44 (2021). Comprehensive review of evidence regarding tumour infiltration by NK cells .
Larsen, S. K., Gao, Y. & Basse, P. H. NK cells in the tumor microenvironment. Crit. Rev. Oncog. 19 , 91–105 (2014).
Cursons, J. et al. A gene signature predicting natural killer cell infiltration and improved survival in melanoma patients. Cancer Immunol. Res. 7 , 1162–1174 (2019).
Borst, L., van der Burg, S. H. & van Hall, T. The NKG2A–HLA-E axis as a novel checkpoint in the tumor microenvironment. Clin. Cancer Res. 26 , 5549–5556 (2020).
Bi, J. & Tian, Z. NK cell exhaustion. Front. Immunol. 8 , 760 (2017).
de Kruijf, E. M. et al. HLA-E and HLA-G expression in classical HLA class I-negative tumors is of prognostic value for clinical outcome of early breast cancer patients. J. Immunol. 185 , 7452–7459 (2010).
Raneros, A. B., Álvarez, B. S. & Larrea, C. L. Secretory pathways generating immunosuppressive NKG2D ligands: new targets for therapeutic intervention. OncoImmunology 3 , e28497 (2014).
Maurer, S. et al. Platelet-mediated shedding of NKG2D ligands impairs NK cell immune-surveillance of tumor cells. OncoImmunology 7 , e1364827 (2018).
Boudreau, J. E. & Hsu, K. C. Natural killer cell education in human health and disease. Curr. Opin. Immunol. 50 , 102–111 (2018).
Björklund, A. T. et al. Complete remission with reduction of high-risk clones following haploidentical NK-cell therapy against MDS and AML. Clin. Cancer Res. 24 , 1834–1844 (2018).
Chen, Z., Yang, Y., Liu, L. L. & Lundqvist, A. Strategies to augment natural killer (NK) cell activity against solid tumors. Cancers 11 , 1040 (2019).
Gauthier, L. et al. Multifunctional natural killer cell engagers targeting NKp46 trigger protective tumor immunity. Cell 177 , 1701–1713.e16 (2019).
Lin, M. et al. Pembrolizumab plus allogeneic NK cells in advanced non-small cell lung cancer patients. J. Clin. Invest. 130 , 2560–2569 (2020).
Zhao, X.-Y. et al. Expanded clinical-grade membrane-bound IL-21/4-1BBL NK cell products exhibit activity against acute myeloid leukemia in vivo. Eur. J. Immunol. 50 , 1374–1385 (2020).
Rock, K. L., York, I. A., Saric, T. & Goldberg, A. L. Protein degradation and the generation of MHC class I-presented peptides. Adv. Immunol. 80 , 1–70 (2002).
Craiu, A., Akopian, T., Goldberg, A. & Rock, K. L. Two distinct proteolytic processes in the generation of a major histocompatibility complex class I-presented peptide. Proc. Natl Acad. Sci. USA 94 , 10850–10855 (1997).
Wearsch, P. A. & Cresswell, P. The quality control of MHC class I peptide loading. Curr. Opin. Cell Biol. 20 , 624–631 (2008).
Fisette, O., Wingbermühle, S., Tampé, R. & Schäfer, L. V. Molecular mechanism of peptide editing in the tapasin–MHC I complex. Sci. Rep. 6 , 19085 (2016).
Thomas, C. & Tampé, R. Proofreading of peptide–MHC complexes through dynamic multivalent interactions. Front. Immunol. 8 , 65 (2017).
Niu, B. et al. MSIsensor: microsatellite instability detection using paired tumor-normal sequence data. Bioinformatics 30 , 1015–1016 (2014).
Chauvin, J.-M. & Zarour, H. M. TIGIT in cancer immunotherapy. J. Immunother. Cancer 8 , e000957 (2020).
Sanchez-Correa, B. et al. DNAM-1 and the TIGIT/PVRIG/TACTILE axis: novel immune checkpoints for natural killer cell-based cancer immunotherapy. Cancers 11 , 877 (2019).
Krijgsman, D., Roelands, J., Hendrickx, W., Bedognetti, D. & Kuppen, P. J. K. HLA-G: a new immune checkpoint in cancer. Int. J. Mol. Sci. 21 , 4528 (2020).
Coulie, P. G., Van den Eynde, B. J., van der Bruggen, P. & Boon, T. Tumour antigens recognized by T lymphocytes: at the core of cancer immunotherapy. Nat. Rev. Cancer 14 , 135–146 (2014). Review on different types of tumour antigen relevant to cancer immunotherapy .
Chiappinelli, K. B. et al. Inhibiting DNA methylation causes an interferon response in cancer via dsRNA including endogenous retroviruses. Cell 162 , 974–986 (2015).
Sheng, W. et al. LSD1 ablation stimulates anti-tumor immunity and enables checkpoint blockade. Cell 174 , 549–563.e19 (2018).
Smith, C. C. et al. Endogenous retroviral signatures predict immunotherapy response in clear cell renal cell carcinoma. J. Clin. Invest. 128 , 4804–4820 (2018).
Kong, Y. et al. Transposable element expression in tumors is associated with immune infiltration and increased antigenicity. Nat. Commun. 10 , 5228 (2019).
Takahashi, Y. et al. Regression of human kidney cancer following allogeneic stem cell transplantation is associated with recognition of an HERV-E antigen by T cells. J. Clin. Invest. 118 , 1099–1109 (2008).
Schumacher, T. N. & Schreiber, R. D. Neoantigens in cancer immunotherapy. Science 348 , 69–74 (2015).
Capietto, A.-H., Jhunjhunwala, S. & Delamarre, L. Characterizing neoantigens for personalized cancer immunotherapy. Curr. Opin. Immunol. 46 , 58–65 (2017).
Vormehr, M., Türeci, Ö. & Sahin, U. Harnessing tumor mutations for truly individualized cancer vaccines. Annu. Rev. Med. 70 , 395–407 (2019).
Burchell, J. M., Mungul, A. & Taylor-Papadimitriou, J. O-linked glycosylation in the mammary gland: changes that occur during malignancy. J. Mammary Gland Biol. Neoplasia 6 , 355–364 (2001).
Cobbold, M. et al. MHC class I-associated phosphopeptides are the targets of memory-like immunity in leukemia. Sci. Transl Med. 5 , 203ra125 (2013).
Vlad, A. M., Kettel, J. C., Alajez, N. M., Carlos, C. A. & Finn, O. J. MUC1 immunobiology: from discovery to clinical applications. Adv. Immunol. 82 , 249–293 (2004).
Borst, J., Ahrends, T., Bąbała, N., Melief, C. J. M. & Kastenmüller, W. CD4 + T cell help in cancer immunology and immunotherapy. Nat. Rev. Immunol. 18 , 635–647 (2018).
Oh, D. Y. et al. Intratumoral CD4 + T cells mediate anti-tumor cytotoxicity in human bladder cancer. Cell 181 , 1612–1625.e13 (2020).
Seliger, B., Kloor, M. & Ferrone, S. HLA class II antigen-processing pathway in tumors: molecular defects and clinical relevance. Oncoimmunology 6 , e1171447 (2017).
Axelrod, M. L., Cook, R. S., Johnson, D. B. & Balko, J. M. Biological consequences of MHC-II expression by tumor cells in cancer. Clin. Cancer Res. 25 , 2392–2402 (2019).
Reizis, B. Plasmacytoid dendritic cells: development, regulation, and function. Immunity 50 , 37–50 (2019).
Bosteels, C. et al. Inflammatory type 2 cDCs acquire features of cDC1s and macrophages to orchestrate immunity to respiratory virus infection. Immunity 52 , 1039–1056.e9 (2020).
Guilliams, M. et al. Unsupervised high-dimensional analysis aligns dendritic cells across tissues and species. Immunity 45 , 669–684 (2016).
Shortman, K. Dendritic cell development: a personal historical perspective. Mol. Immunol. 119 , 64–68 (2020).
Cohn, L. et al. Antigen delivery to early endosomes eliminates the superiority of human blood BDCA3 + dendritic cells at cross presentation. J. Exp. Med. 210 , 1049–1063 (2013).
Zelenay, S. et al. The dendritic cell receptor DNGR-1 controls endocytic handling of necrotic cell antigens to favor cross-priming of CTLs in virus-infected mice. J. Clin. Invest. 122 , 1615–1627 (2012).
Theisen, D. J. et al. WDFY4 is required for cross-presentation in response to viral and tumor antigens. Science 362 , 694–699 (2018).
Bennett, S. R. M. et al. Help for cytotoxic-T-cell responses is mediated by CD40 signalling. Nature 393 , 478–480 (1998).
Schoenberger, S. P., Toes, R. E. M., van der Voort, E. I. H., Offringa, R. & Melief, C. J. M. T-cell help for cytotoxic T lymphocytes is mediated by CD40–CD40L interactions. Nature 393 , 480–483 (1998).
Boegel, S. et al. HLA and proteasome expression body map. BMC Med. Genomics 11 , 36 (2018).
Morozov, A. V. & Karpov, V. L. Proteasomes and several aspects of their heterogeneity relevant to cancer. Front. Oncol. 9 , 761 (2019).
Fabre, B. et al. Label-free quantitative proteomics reveals the dynamics of proteasome complexes composition and stoichiometry in a wide range of human cell lines. J. Proteome Res. 13 , 3027–3037 (2014).
Guillaume, B. et al. Two abundant proteasome subtypes that uniquely process some antigens presented by HLA class I molecules. Proc. Natl Acad. Sci. USA 107 , 18599–18604 (2010).
Guillaume, B. et al. Analysis of the processing of seven human tumor antigens by intermediate proteasomes. J. Immunol. 189 , 3538–3547 (2012).
Download references
Author information
Authors and affiliations.
Genentech Inc., South San Francisco, CA, USA
Suchit Jhunjhunwala, Christian Hammer & Lélia Delamarre
You can also search for this author in PubMed Google Scholar
Contributions
L.D. and S.J. contributed equally to the manuscript as a whole. C.H. led the NK cell topic.
Corresponding authors
Correspondence to Suchit Jhunjhunwala or Lélia Delamarre .
Ethics declarations
Competing interests.
All the authors are employees of Genentech Inc.
Additional information
Peer review information.
Nature Reviews Cancer thanks M. Smyth, S. Spranger and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Related links
cBioportal: https://www.cbioportal.org/
Supplementary information
Supplementary information.
(MHC). A locus that encodes several genes involved in antigen presentation and other related immune processes.
(HLA). In humans, MHC is also called HLA. HLA-I, or MHC class-I, includes classical HLA-Ia genes ( HLA-A , HLA-B and HLA-C ) and non-classical HLA-Ib genes. Classical HLA genes present peptides at the cell surface, while non-classical HLA gene products have several other functions including natural killer cell activation or inhibition, and presentation of metabolites, lipids, etc. In this Review, we use HLA-I to refer to the classical genes only. Similarly, HLA-II will be used to refer to classical MHC class-II genes. For most of the discussion, we use the term HLA instead of MHC.
Mutated peptides presented on the tumour cell surface by HLA. They are specific to tumours, as they arise from somatic mutations, thus distinguishing them from self antigens.
(pAPCs). Cells that specialize in presenting antigens on MHC molecules to prime and stimulate T cells. These include dendritic cells, macrophages and B cells.
(APM). Includes the peptide loading complex and also peptide processing machinery such as the proteasome.
The delivery of an innate stimulus to dendritic cells (DCs) at the tumour site. Unlike conventional vaccines, which co-deliver antigens and innate stimulus to DCs to stimulate antitumour T cell immunity, in situ vaccines rely on the antigens released by dying tumour cells as a source of tumour antigens for DCs. Examples of innate stimuli evaluated in the clinic are TLR agonists (TLR7/8 ligands, TLR9 ligands, the TLR3 ligand poly(I:C)), STING agonist and anti-CD40 agonist antibody.
(PLC). Includes the core set of proteins in the endoplasmic reticulum (ER) that mediate peptide transport into the ER and subsequent loading of peptide onto HLA-I. These include TAP1, TAP2, tapasin, ERp57, calnexin, calreticulin, ERAP1, ERAP2, HLA-I and β2m.
HLA is the most polymorphic locus in humans, with more than 19,000 alleles documented. HLA-I consists of three genes, and since both alleles of each gene are expressed, up to six different HLA-I proteins or allotypes may be expressed in an individual, with each allotype presenting its own set of peptides. As different HLA-I allotypes may present a distinct set of peptides, the total repertoire of peptides presented by HLA-I (also called the HLA-I ligandome) is highly diverse.
A catabolic pathway that degrades cytosolic components including proteins and organelles. Autophagosomes capture these cytosolic materials and fuse with lysosomes to mediate their degradation.
ERAP1 and ERAP2 are endoplasmic reticulum-resident aminopeptidases that may trim peptides that bind to HLA-I. ERAP1 and ERAP2 are collectively referred to as ERAP.
Rights and permissions
Reprints and permissions
About this article
Cite this article.
Jhunjhunwala, S., Hammer, C. & Delamarre, L. Antigen presentation in cancer: insights into tumour immunogenicity and immune evasion. Nat Rev Cancer 21 , 298–312 (2021). https://doi.org/10.1038/s41568-021-00339-z
Download citation
Accepted : 01 February 2021
Published : 09 March 2021
Issue Date : May 2021
DOI : https://doi.org/10.1038/s41568-021-00339-z
Share this article
Anyone you share the following link with will be able to read this content:
Sorry, a shareable link is not currently available for this article.
Provided by the Springer Nature SharedIt content-sharing initiative
This article is cited by
Il-2rg as a possible immunotherapeutic target in crc predicting poor prognosis and regulated by mir-7-5p and mir-26b-5p.
- Ehsan Gharib
- Leili Rejali
- Ehsan Nazemalhosseini-Mojarad
Journal of Translational Medicine (2024)
Phylogenetic analysis and antigenic epitope prediction for E6 and E7 of Alpha-papillomavirus 9 in Taizhou, China
BMC Genomics (2024)
The oncolytic bacteria-mediated delivery system of CCDC25 nucleic acid drug inhibits neutrophil extracellular traps induced tumor metastasis
- Zi-chun Hua
Journal of Nanobiotechnology (2024)
Crosstalk between colorectal CSCs and immune cells in tumorigenesis, and strategies for targeting colorectal CSCs
- Shuiling Jin
Experimental Hematology & Oncology (2024)
Functional CRISPR screens in T cells reveal new opportunities for cancer immunotherapies
- Minghua Xiang
Molecular Cancer (2024)
Quick links
- Explore articles by subject
- Guide to authors
- Editorial policies
Sign up for the Nature Briefing: Cancer newsletter — what matters in cancer research, free to your inbox weekly.


FDA announces firm distributing Chinese-made plastic syringes in the U.S. initiates recall

The Food and Drug Administration May 23 announced that Medline Industries, a firm marketing and distributing Chinese-manufactured plastic syringes within the U.S., initiated a recall to stop using affected products, including unauthorized plastic syringes made in China. On Tuesday, Chinese manufacturer Jiangsu Shenli Medical Production Co. initiated a recall to stop using its unauthorized syringes. The FDA last week recommended users immediately transition away from plastic syringes made by Jiangsu Shenli Medical Production Co., along with those made by Jiangsu Caina Medical Co., Zhejiang Longde Pharmaceutical Co. and Shanghai Kindly Enterprise Development Group Co.
Related News Articles
IMAGES
VIDEO
COMMENTS
Antigen presentation is a vital immune process that is essential for T cell immune response triggering. Because T cells recognize only fragmented antigens displayed on cell surfaces, antigen processing must occur before the antigen fragment can be recognized by a T-cell receptor. Specifically, the fragment, bound to the major histocompatibility ...
Antigen Processing and Presentation. In order to be capable of engaging the key elements of adaptive immunity (specificity, memory, diversity, self/nonself discrimination), antigens have to be processed and presented to immune cells. Antigen presentation is mediated by MHC class I molecules, and the class II molecules found on the surface of ...
Abstract. Antigen processing and presentation are the cornerstones of adaptive immunity. B cells cannot generate high-affinity antibodies without T cell help. CD4 + T cells, which provide such ...
Antigen Processing and Presentation. T cells can only recognise antigens when they are displayed on cell surfaces. This is carried out by Antigen-presenting cells (APCs), the most important of which are dendritic cells, B cells, and macrophages. APCs can digest proteins they encounter and display peptide fragments from them on their surfaces ...
Antigen presentation is a process in the body's immune system by which macrophages, dendritic cells and other cell types capture antigens, then present them to naive T-cells. The basis of adaptive immunity lies in the capacity of immune cells to distinguish between the body's own cells and infectious pathogens. The host's cells express ...
An antigen-presenting cell (APC) is an immune cell that detects, engulfs, and informs the adaptive immune response about an infection. When a pathogen is detected, these APCs will phagocytose the pathogen and digest it to form many different fragments of the antigen. Antigen fragments will then be transported to the surface of the APC, where ...
15.4M: Antigen Presentation. Antigens are macromolecules that elicit an immune response in the body. Antigens can be proteins, polysaccharides, conjugates of lipids with proteins (lipoproteins) and polysaccharides (glycolipids). Most of this page will describe how protein antigens are presented to the immune system.
Antigen Presentation and Major Histocompatibility Complex. Pavel P. Nesmiyanov, in Encyclopedia of Infection and Immunity, 2022 Abstract. Antigen presentation is a process that allows T cells to recognize antigenic epitopes displayed on the surface of an antigen-presenting cell. Antigen presentation involves a sophisticated process of epitope preparation, i.e., processing which involves ...
ANTIGEN PRESENTATION. In Immunology Guidebook, 2004. ANTIGEN PRESENTATION. Antigen presentation is the expression of antigen molecules on the surface of a macrophage or other antigen-presenting cell in association with MHC class II molecules when the antigen is being presented to a CD4 + helper T cell or in association with MHC class I molecules when presentation is to CD8 + cytotoxic T cells.
Definition. Antigen presentation is the process by which protein antigen is presented to lymphocytes in the form of short peptide fragments. These are associated with antigen-presenting molecules ...
An antigen-presenting cell ( APC) or accessory cell is a cell that displays an antigen bound by major histocompatibility complex (MHC) proteins on its surface; this process is known as antigen presentation. T cells may recognize these complexes using their T cell receptors (TCRs). APCs process antigens and present them to T cells.
Peter Cresswell's review (Cresswell 2019) is a personal retrospective focusing on the biochemistry of antigen presentation. His seminal work on class I and class II processing, as well as on cross-priming, are written like detective stories, recounting how he and his colleagues progressed step by step in deciphering the pathways defining ...
Avian Antigen-Presenting Cells. Bernd Kaspers, Pete Kaiser, in Avian Immunology (Second Edition), 2014. 9.1.1 Antigen Presentation. Antigen presentation is the mechanism by which the antigenic environment is sampled and information imparted to the effector arms of the adaptive immune system, B and T lymphocytes. Depending on the precise context, antigen presentation can result in either ...
Antigen presentation lies at the heart of immune recognition of infected or malignant cells. For this reason, important efforts have been made to predict which peptides are more likely to bind and be presented by the human leukocyte antigen (HLA) complex at the surface of cells.
Antigen Processing and Presentation. Antigen processing and presentation is the process of digestion of antigens into smaller peptide fragments by an antigen-presenting cell (APCs) that are then displayed on the surface of the cells via antigen-presenting molecules like MHC class I and II for recognition by lymphocytes.. Antigen processing and presentation can occur via three different pathways;
Abstract | Antigen processing and presentation are the cornerstones of adaptive immunity. B cells cannot generate high- affinity antibodies without T cell help. CD4+ T cells, which provide such ...
Professional antigen presenting cells (APCs) are immune cells that specialize in presenting an antigen to a T-cell. The main types of professional APCs are dendritic cells (DC), macrophages, and B cells. A professional APC takes up an antigen, processes it, and returns part of it to its surface, along with a class II major histocompatibility ...
Direct Antigen Presentation. Dendritic cells, monocytes, macrophages and B cells can all act as antigen-presenting cells (APC). Dendritic cells are the most important APC because they are most efficient at taking up antigens and processing them for presentation to T cells. Tumor cells that express antiben bound to MHC class I can also be ...
Although this is a key step in antigen cross-presentation and significant efforts have been made to shed light on this process, the underlying mechanisms mediating such intracellular antigen transport are still topic of debate. ... Schaefer M, Tannert A, et al. Superior antigen cross-presentation and XCR1 expression define human CD11c+CD141 ...
Antigen processing and presentation is the mechanism by which whole antigens are degraded and loaded onto MHC molecules for display on the cell surface for recognition by T cells. Both macrophages ...
Figure 1. Antigen presentation on MHC-II molecule. Extracellular antigens are endocytosed or phagocytosed, and intracellular antigens are translocated to the late-endosome or the lysosome via autophagosome- or LAMP-2A- mediated autophagy. Then these antigens are degraded by asparaginyl endopeptidase and cathepsin.
Types of Antigen-Presenting Cells. Dendritic Cells. Location: Found in tissues that are in contact with the external environment, such as the skin (Langerhans cells), and the inner lining of the nose, lungs, stomach, and intestines. Function: Capture antigens, migrate to lymph nodes, and present them to T cells.
MHC-I antigen presentation. Additionally, our work introduces an approach to quantify and visualize defined pMHCI - complexes, opening broad avenues for immunological research. Competing interest statement: B.W. and D.K.C. indicate a potential conflict of interest as former and present employee of Immunocore Ltd, respectively. The remaining
MHC complexes. Antigen processing and presentation enable the adaptive immune system to survey the host cell proteome and detect pathogens and mutations 36,37.MHC I and MHC II are the two ...
During the presentation, preliminary progression-free survival and overall survival data will be shared for the first time. ... engineered to induce robust and durable antigen-specific CD8+ T cell ...
The difference between wild-type and 5xFAD mice was differential according to the definition (by gene combination to define reactivity) of reactive astrocytes and reactive microglia in their density and distribution (Fig. 2 and Supplementary Fig. 7).Reactive astrocytes and reactive microglia shared gene signatures and were supposed to collaborate to do the job of waste disposal in situ and out ...
We further explored the key genes associated with antigen presentation to identify valuable predictive markers. Since the KEGG antigen processing and presentation (APM) pathway was indicated to be enriched in responders through GSEA analysis, we focused on the core enriched genes of this pathway. ... The bar of scatterplot indicates the mean ...
There is a growing body of studies focusing on the identification of novel autoantibodies and the definition of their clinical implications, ... with particular emphasis on processes linked to antigen presentation and immune cell activation; (2) we showed the increased production of profibrotic cytokines, including IL-4, IL-13, IL-6 and IL-1β ...
Immune checkpoint inhibition does not benefit all patients. This Review discusses how antigen presentation, which is crucial for the success of this therapy, may be disrupted in tumours and ...
The Food and Drug Administration May 23 announced that Medline Industries, a firm marketing and distributing Chinese-manufactured plastic syringes within the U.S., initiated a recall to stop using affected products, including unauthorized plastic syringes made in China. On Tuesday, Chinese manufacturer Jiangsu Shenli Medical Production Co. initiated a recall to stop using its unauthorized ...