- Open access
- Published: 16 May 2022

Cystic fibrosis transmembrane conductance regulator (CFTR): beyond cystic fibrosis
- Giuseppe Fabio Parisi ORCID: orcid.org/0000-0003-4291-0195 1 ,
- Federico Mòllica ORCID: orcid.org/0000-0001-8417-9201 1 ,
- Alessandro Giallongo ORCID: orcid.org/0000-0002-7966-5800 1 ,
- Maria Papale ORCID: orcid.org/0000-0001-5036-2374 1 ,
- Sara Manti ORCID: orcid.org/0000-0002-7664-3083 1 &
- Salvatore Leonardi ORCID: orcid.org/0000-0002-2642-6400 1
Egyptian Journal of Medical Human Genetics volume 23 , Article number: 94 ( 2022 ) Cite this article
3719 Accesses
5 Citations
Metrics details
The cystic fibrosis transmembrane conductance regulator ( CFTR ) gene has been traditionally linked to cystic fibrosis (CF) inheritance in an autosomal recessive manner. Advances in molecular biology and genetics have expanded our understanding of the CFTR gene and its encoding products expressed in different tissues.
The study’s aim consists of reviewing the different pathological CF phenotypes using the existing literature. We know that alterations of the CFTR protein’s structure may result in different pathological phenotypes.
Open sources such as PubMed and Science Direct databases have been used for this review. We focused our selection on articles published within the last 15 years. Critical terms related to the CFTR protein have been used: “CFTR AND cancer,” “CFTR AND celiac disease,” “CFTR AND pancreatitis,” “children,” “adults,” “genotype,” “phenotype,” “correlation,” “mutation,” “CFTR,” “diseases,” “disorders,” and “no cystic fibrosis.”
We analyzed 1,115 abstracts in total. Moreover, only 189 were suitable for the topic. We focused on the role of CFTR in cancer, gastrointestinal disorders, respiratory diseases, reproductive system, and systemic hypertension.
Conclusions
Mutations in CFTR gene are often associated with CF. In this review, we highlighted the broad spectrum of alterations reported for this gene, which may be involved in the pathogenesis of other diseases. The importance of these new insights in the role of CFTR relies on the possibility of considering this protein/gene as a novel therapeutic target for CF- and CFTR-related diseases.
Introduction
The gene that encodes the human cystic fibrosis transmembrane conductance regulator ( CFTR ) protein is located on the long arm of chromosome 7. It encodes for a membrane protein, specifically the ATP-binding cassette transporter-class ion channel protein that conducts chloride and thiocyanate ions across epithelial cell membranes. Mutations in the CFTR gene result in cystic fibrosis (CF), an autosomal recessive disorder. Since its discovery in 1989, over 2,300 variations are reported for the CFTR gene [ 1 , 2 ].
Genetic testing availability and accessibility have broadened the spectrum of genotypes related to CFTR and the genotype–phenotype correlation, especially for milder phenotypes [ 3 , 4 , 5 ]. CFTR is expressed in different tissues and organs, mainly lung, gastrointestinal, and reproductive systems. Mutations affecting chloride ion channel function might lead to dysregulation of epithelial fluid transport in the lung and pancreas and affect other organs [ 6 , 7 ].
New evidence has, recently, reconsidered the role of CFTR in diseases other than cystic fibrosis [ 8 ]. CFTR dysfunctions have been reported in high-heavy diseases such as cancer, celiac disease, and chronic obstructive pulmonary disease (COPD) [ 8 ]. Unlike CF, the suggested underlying pathogenic mechanisms include heterozygous mutations and an acquired dysfunction, such as epigenetic mechanisms [ 8 ]. This study reviews the existing literature on the different pathological phenotypes other than CF caused by alterations to the CFTR protein.
Research strategy
Open sources such as PubMed and Science Direct databases have been used for this review. We focused our selection on articles published within the last 15 years. Critical terms related to the CFTR protein have been used: “CFTR AND cancer,” “CFTR AND celiac disease,” “CFTR AND pancreatitis,” “children,” “adults,” “genotype,” “phenotype,” “correlation,” “mutation,” “CFTR,” “diseases,” “disorders,” and “no cystic fibrosis.”
Study selection
The following inclusion criteria have been considered in the article selection: English language, publication in peer-reviewed journals, published since 2006. All the articles irrelevant to the investigated issue have been excluded by title, abstract, or full text. Articles concerning cystic fibrosis were excluded. The studies containing inclusion criteria in the abstract have been considered for clarification. The selection has been extended to article’s references with similar inclusion criteria. A final number of 189 of 1,115 abstracts have satisfied the inclusion criteria.
CFTR and cancer
A growing number of studies have recently highlighted a correlation between mutations in the CFTR gene and different types of cancers [9–36, Table 1 ]. The role of CFTR in cancer seems to be variable according to the neoplasm, probably due to the influence and interaction with different tissue microenvironments [ 9 ].
A large population-based study on around 500,000 individuals assessed the association between CFTR mutation carriers, specifically F508del, and the risk of 54 types of cancers using the United Kingdom Biobank data [ 10 ]. Compared to non-cancer subjects, a significantly higher CFTR F508del mutation rate was found in individuals affected by colorectal ((OR 1.17 (95% CI 1.02–1.32, p = 0.02)), gallbladder and biliary tract ((OR 1.92 (95% CI 1.20–2.91, p = 0.004)), thyroid cancer ((OR 1.47 (95% CI 0.99–2.08, p = 0.04)), and non-Hodgkin's lymphoma ((OR 1.32 (95% CI 1.04–1.65, p = 0.02)) [ 10 ], and remained significantly high after multivariable analysis. Overall, lung cancer risk was reduced; however, in a similar study on Danish CFTR F508del, an increased risk of lung cancer was observed ((OR 1.52 (95% CI 1.12–2.08, p = 0.008)) [ 11 ]. No significant association between mutations and pancreatic cancer ((OR 1.2 (95% CI 0.85–1.64)) was observed [ 10 ]. This was consistent with Schubert et al. about pancreatic cancer, though they had a smaller study of 31 different CFTR mutations [ 12 ]. Nevertheless, a meta-analysis identified a modest but significantly increased risk of pancreatic cancer in four of the 13 CFTR mutation carriers (OR 1.41, 95% CI 1.07–1.84, p = 0.013, [mutations: F508del, W1282X, ΔI507, S549R]) [ 13 ]. Thus, this difference might be attributable to the spectrum of mutations analyzed.
The role of CFTR in cancer pathogenesis is not limited to gene mutations, but also through epigenetic modifications [ 14 ]. It has been suggested that the expression of CFTR was downregulated in patients affected by esophageal cancer ( p < 0.05). Indeed, CFTR overexpression inhibited the growth and migration of esophageal cancer cells by downregulating protein expression of the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) ( p < 0.05). Conversely, CFTR silencing led to an increase in NF-κB-p65 and -p50 expression and increased tumor invasion, and growth in mice. [ 15 ].
Similarly, in colorectal cancer, CFTR expression was downregulated compared to normal tissues ( p = 0.034), possibly due to the CFTR promoter's methylation. Interestingly, CFTR overexpression acts as a tumor suppressor and reduces cell migration and invasion [ 16 ], possibly through increased Wnt/beta-catenin signaling evidenced in CFTR knock-out mice, though this data remains to be verified [ 17 ]. Liu et al. found that knocked down CFTR colorectal cancer cells reduced heat shock protein 90 (HSP-90) expression ( p < 0.01), leading to altered AKT and extracellular-regulated kinase (ERK) signaling pathways. Reduced AKT ERK signaling pathways determined mitochondrial dysfunction by inhibiting the Bcl-2 family proteins involved in apoptosis [ 18 ].
Hypermethylation of the CFTR promoter is observed in head and neck cancer and non-small cell lung, bladder, hepatic/hepatocellular, and breast cancer [ 19 , 20 , 21 , 22 , 23 ]. In the latter, CFTR silencing may lead to loss of E-cadherin, which assists in cell adhesion. The loss of E-cadherin correlates with invasion and metastasis [ 20 , 24 ].
CFTR is expressed in the epithelial cells along the female reproductive tract [ 25 ]. CFTR overexpression in ovarian cancer is positively associated with severe cancers, at an advanced stage, and have a higher degree of malignancy, based on 112 tissue samples (83 epithelial ovarian cancer) ( p < 0.05). The authors suggested a possible interaction with intracellular c-Src signaling pathways involved in cellular growth [ 26 ]. A similar observation concerning CFTR overexpression was reported in cervical cancer ( p < 0.01), where it was associated with poorer prognosis, stage and metastasis. [ 27 ]. However, a more recent study contradicted these results, reporting that CFTR overexpression mediated the inhibition of the NF-κB p65 signaling pathway and reduced cell proliferation and tumor invasion [ 28 ]. In endometrial carcinoma, increased CFTR mRNA expression was identified ( p < 0.05). Nevertheless, inhibition of CFTR with the CFTR inhibitor 172 promoted cell proliferation through reduced micro-RNA (miR)-125b, which acts as a tumor suppressor, decreasing the expression of matrix metalloproteinase 11 ( MMP11 ), and vascular endothelial growth factor (VEGF) −A ( p < 0.05) [ 29 ]. Further, high levels of urokinase plasminogen activator (uPA) are observed in prostate cancer, which leads to cell proliferation. Interestingly, a high association between CFTR expression and miR-193b expression was identified in those with prostate cancer. A reduction in CFTR expression leads to a reduction in miR-193A, with subsequent lack of inhibition of its target, the uPA [ 30 ]. Thus, these findings provide evidence for the many different interactions between CFTR and different signaling pathways, which goes beyond the original role of CFTR as an ion channel only.
The importance of detailed genetic characterization in cancer might have implications for diagnosis, prognosis, and treatment. Tu et al. identified CFTR downregulation in 225 cases of nasopharyngeal cancer. After multivariate analysis, CFTR downregulation resulted as an independent prognostic factor ( p = 0.003), correlating with advanced cancer stages ( p = 0.026), increased metastasis ( p < 0.001), and poor prognosis ( p < 0.01) [ 31 ]. Although lower CFTR expression was associated with a poor prognosis in 153 patients with glioblastoma ( p = 0.04) [ 32 ], increased CFTR expression was also not beneficial to prostate cancer prognosis because it correlated with chemotherapy resistance [ 33 ]. However, Xie et al. reported that CFTR overexpression, whose was downregulated in prostate cancer by hypermethylation, suppressed prostate cancer progression in vitro and in vivo [ 30 ]. The emerging role of CFTR in cancer represents an intriguing potential therapeutic target. Indeed, like Forskolin and Cact-A1, CFTR activators demonstrated an antiproliferative effect on glioblastoma by significantly reducing Ki67 positive cells ( p < 0.05) [ 32 ]. CFTR activation may decrease phosphorylation of Janus kinase 2/signal transducer and activator of transcription-3 (JAK2/STAT3) signaling pathway. This effect was attenuated by CFTR -inh172 ( p < 0.05) [ 32 , 33 ]. CFTR overexpression exerted a similar influence on lung adenocarcinoma Ki67 positive cells, as well as on cells’ invasion and migration. CFTR overexpression was also effective to inhibit clonogencity induced by nicotine exposure ( p < 0.01) [ 34 ].
In contrast to this, in diseases where the Philadelphia chromosome is present, leading to T-cell acute lymphoblastic leukemia, CFTR expression was higher than normal controls ( p < 0.001), Philadelphia chromosome negative acute lymphoblastic leukemia and chronic myeloid leukemia cells) ( p < 0.01). Further, using the CFTR inhibitor, CFTR -inh172, inhibited T-cell acute lymphoblastic leukemia growth. The underlying mechanism of cell proliferation in this subtype of leukemia involved activating the p-BCR-ABL and Wnt/β-catenin signaling pathway. This leads to constant CFTR activation, which mediated inhibition of protein phosphatase 2A (PP2A) antitumoral activity [ 35 , 36 ].
In summary, studies on cancer have overcome the conception of CFTR as a mere ion channel and transporter but showing pleiotropic interactions with proteins from different intracellular signaling pathways. The disruption of these pathways is from the consequence of gene mutations and the result of epigenetic modifications.
CFTR and gastrointestinal disorders
The most common gastrointestinal disorder associated with CFTR mutations is pancreatic insufficiency and pancreatitis in the setting of CF [ 37 ]. However, a fourfold increased risk of CFTR mutation carriers in idiopathic chronic pancreatitis (ICP) sufferers also has emerged, though the small sample size (OR 4.3, 95% CI 2.1–8.7, p = 0.0002). Further, Cohn extended the analysis to three previous studies. Overall, 14 CFTR mutation carriers out of 155 individuals with ICP were found, and the OR was 2.9 (1.7–4.9, p < 0.0001) [ 38 ]. The p.R75Q CFTR heterozygous variant is associated with ICP (OR = 3.43, 95% CI 1.7–6.8). The risk for ICP was much higher with the serine peptidase inhibitor ( SPINK ) 1 heterozygous variant (OR = 62.5, 95% CI 16.6–95.4). Other CFTR variants, except for F508del, became significant only when inherited together with SPINK1 variants, which may be due to CFTR impaired HCO 3 − conductance and not chloride conductance in p.R75Q carriers. Overall, CFTR carriers, including CF-causing variants, had an OR of 7.4 (95% CI 2.3–18.5) of those with ICP [ 39 ]. Two previous review study partially reconsidered these results, highlighting the predominant role of SPINK1 in those with ICP when non-CF-causing variants, such as p.R75Q, were detected. Their rate was not significantly different compared to controls carrying SPINK1 variants [ 40 , 41 ].
Beyond CFTR , the most common genes associated with ICP were the cationic trypsinogen gene ( PRSS1 ), SPINK1 , and CTRC gene. In a Chinese cohort of 715 individuals with ICP, isolated CFTR variants were not significantly associated with ICP compared with 1,196 controls ( p > 0.05) [ 42 ]. Therefore, although the association between CFTR and pancreatitis has been widely investigated, there are still contradictory results due to the complex genetic background and interactions between environmental factors.
Other gastrointestinal disorders should be added within the spectrum of the CFTR gene mutations and dysfunctions. These include secretory diarrheas, altered bile acid homeostasis, primary sclerosing cholangitis, and celiac disease [ 43 ]. CFTR targets bacterial enterotoxins on the intestinal epithelial cell membrane, which induce cAMP- or cGMP-mediated protracted activation of CFTR , leading to subsequent water loss and dehydration, typical features of secretory diarrheas. Thus, CFTR inhibitors, like CFTR -inh172, could represent a potential target therapy in secretory diarrheas. However, some limitations include the difficult localization of CFTR in the intestinal crypts and other ion channels' involvement in the pathogenesis of secretory diarrheas [ 43 , 44 ].
Another organ affected by CFTR dysfunction is the gallbladder, as evidenced in CFTR knock-out mice, which showed impaired gallbladder emptying ( p < 0.05), probably due to vasoactive intestinal peptide (VIP) overexpression which acts as a myorelaxant on gallbladder, and bile acid homeostasis [ 44 ]. Primary sclerosing cholangitis (PSC) is a chronic cholestasis condition associated with biliary inflammation, obliteration, and fibrosis [ 45 ]. Though PSC pathogenesis has been associated with specific HLAs (HLA-DRB1*1501-DQB1*0602, HLA-DRB1*1301-DQB1*0603, and HLA-A1-B8-DRB1*0301-DQB1*0201) and often with inflammatory bowel disease, a role for CFTR in PSC development has been suggested [ 46 , 47 ]. Sheth et al. reported a significant increase in heterozygous CFTR variants in seven out of 19 (37%, 95% CI: 16–62%) individuals affected by PSC compared to inflammatory bowel disease and primary biliary cirrhosis patients ( p < 0.02) [ 47 ]. In a cohort of 32 patients with PSC who underwent next-generation sequencing, six had CFTR disease causing mutations on one allele (OR = 6.1–95% CI 2.2–16.7, p = 0.002), and 19 carried at least one CFTR polymorphism. However, six had abnormal, and 21 had intermediate sweat tests associated with CF-like phenotype [ 48 ]. Previous studies questioned the relationship between CFTR and PSC, which did not find significant differences between patients with or without PSC and the prevalence of CFTR mutations. Nevertheless, these studies may be biased by the limited number of modifications analyzed [ 48 , 49 ].
Celiac disease (CD) is strictly associated with HLA DQ2/DQ8 predisposition [ 50 ]. The evidence that CF patients reported having a significantly higher CD incidence ( p = 0.0007) has led to an investigation of CFTR's function in patients with CD [ 51 , 52 ]. Interestingly, Villella et al. revealed new insights in such a disease's pathogenesis, suggesting a CFTR role as a gluten target [ 53 ]. In previous studies, CFTR has been identified as a regulator of proteostasis, an essential cytoprotective mechanism to remove misfolded or polyubiquitylated proteins. Its dysfunction was associated with lung inflammation due to autophagy inhibition, mediated by tissue transglutaminase 2 (TGM2), a key enzyme involved in CD [ 54 ]. Villella et al. showed that alpha-gliadin-derived LGQQQPFPPQQPY peptide (P31–43) inhibits the ATPase function of CFTR by binding with the nucleotide-binding domain-1 (NBD1) of CFTR on intestinal epithelial cells of mice with CD-predisposing HLA. NBD1 interaction with P31–43 happened only when it is in a closed conformation. This has consequences on downstream pathways, the tissue transglutaminase 2 (TGM2), and the autophagy protein Beclin-1 (BECN1). Indeed, TGM2 activation, in response to CFTR inactivation, led to reduced BECN1 with impaired proteostasis, determining a pro-inflammatory state. Therefore, TGM2, inhibiting NF-κB inhibitor alpha (NFKBIA), determined increased NFκB and inflammasome activation with IL-15 and IL-β production, respectively. These initiate an immune response against gliadin [ 53 ].
The previously mentioned interactions created a vicious cycle that amplifies and further worsen CFTR inhibition. Hence, Maiuri et al. introduced the definition of “infernal trio” regarding CFTR inhibition, TGM2 activation, and autophagy impairment. For example, TGM2 activation promoted CFTR crosslinking with P31-43, creating a trimolecular complex, which made CFTR inhibition irreversible [ 55 ]. In the light of this mechanism, and the affinity of P31-43 peptide to the closed conformation of CFTR, potentiators of CFTR may be a therapeutic option in celiac disease, by promoting CFTR channel opening. VX-770 (Ivacaftor), a CFTR potentiator, seemed to effectively reduce gliadin-induced inflammation in vitro, especially IL-15 production and NF-kB p65, making CFTR a potential new therapeutic target in CD. Furthermore, VX-770 also prevented gliadin mediated inhibition of CFTR and promoted a tolerogenic response in gluten-sensitive mice and cells from celiac patients [ 53 ].
Genistein, a phytoestrogen contained in soy, targets CFTR and acts as a channel gating potentiate. In the context of celiac disease, it was able to prevent P31-43 induced epithelial stress and inflammation, both in vitro and in vivo animal models [ 56 ].
Drugs targeting CFTR may find application in two other autoimmune diseases, idiopathic autoimmune pancreatitis and Sjogren’s syndrome, characterized by disrupted fluid secretion in pancreatic ducts and of saliva and lacrimal glands, respectively. Indeed, in Sjogren's syndrome mice model CFTR expression was reduced and treatment with VX-770 and C18 increased salivation, ductal fluid secretion, and reduced inflammation. Similar results were obtained in a pancreatic inflammation model. Furthermore, C18 restored CFTR expression in the ducts of salivary glands. Interestingly, this had also effects on acinar cells, where Aquaporin 5 expression was recovered [ 57 ]. Table 2 summarizes the main studies about CFTR and gastrointestinal disorders.
CFTR and the lungs
Recently, acquired CFTR dysfunctions was found to occur in highly prevalent diseases such as chronic obstructive pulmonary disease (COPD) with a chronic bronchitis phenotype [ 58 ]. Indeed, cigarette smoke exposure reduces CFTR expression and activity, both in vitro and in vivo, contributing to mucociliary clearance impairment. Interestingly, CFTR dysfunction was not limited to the lungs but was evidenced in other areas, suggesting a potential role in the onset of smoke-related complications such as Mellitus diabetes, male infertility, and idiopathic pancreatitis [ 59 ]. The hypothesized mechanism may be attributable to acrolein, which induces structural changes to the CFTR channel, or cadmium and arsenic [ 59 , 60 , 61 ]. As regards cadmium, it showed to reduce CFTR expression ( p < 0.001), both in vitro and in vivo, in a dose- and time-dependent manner and CFTR channel activity [ 60 ]. A similar effect was described for arsenic through ubiqutin-mediated lysosomal degradation of CFTR and reduced chloride secretion in vitro ( p < 0.05) [ 61 ]. In a double-blind, placebo-controlled study on 92 COPD patients, the CFTR potentiator Icenticaftor 300 mg effectively improved FEV1 after 28 days (mean 50 mL for pre-bronchodilator FEV1 and mean 63 mL for post-bronchodilator FEV1), while no significant improvement was seen in the lung clearance index [ 62 ].
In a population-based study, 2858 carriers of CFTR F508del were identified and had an increased risk of chronic bronchitis (HR 1.31, 95% CI 1.16–1.48), bronchiectasis (HR 1.88, 95% CI 1.03–3.45), and lung cancer (HR 1.52, 95% CI 1.12–2.08). However, this study's main limitation is the lack of investigations for mutations different from F508del, which may be responsible for compound heterozygosis [ 11 ].
Bronchial asthma is certainly one of the main chronic diseases of the respiratory system [ 63 , 64 ]. Although the risk of asthma was not higher than the general population, a meta-analysis of 15 studies (2.113 asthma cases and 13.457 controls) on CFTR mutations carriers, which contained 34 different pathogenetic variants, reported an increased risk of asthma (OR 1.61, 95% CI 1.18–2.21) [ 65 ]. Table 3 summarizes the main studies about CFTR and lung disorders.
CFTR and the reproductive system
Congenital bilateral absence of the vas deferens (CBAVD) accounts for approximately 3% of infertility cases. Because almost all infertile CF males exhibit CBAVD, it is widely considered an atypical form of CF and a CFTR-related disorder [ 7 ]. Interestingly, a meta-analysis on 38 studies found that 28% (95% CI 24–32%) of individuals with CBAVD carried only one CFTR mutation. However, there is a considerable risk of bias because of the included study's heterogeneity (Egger’s test p = 0.874), which might not have investigated less common CFTR [ 66 ]. Recently, heterozygous copy number variants of CFTR have also been suggested to play a role in the pathogenesis of CBAVD, reported to affect five (1.9%) of 263 Chinese affected individuals. Among these, four out of five carried a CFTR partial deletion [ 67 ].
Regarding the female reproductive system, in polycystic ovarian syndrome (PCOS), CFTR and aromatase expression was downregulated in granulosa cells, both in rat model and human, compared with non-PCOS women ( p < 0.05). Hence, Chen et al. found that CFTR enhanced follicle-stimulating hormone (FSH) mediated aromatase expression through HCO 3 -induced cAMP response element-binding protein (CREB) phosphorylation [ 68 ]. In a more recent study, the CFTR/HCO 3 -/sAC signaling pattern was further elucidated, contributing to MAPK/ERK downstream pathways of cell proliferation. Therefore, defective CFTR decreased granulosa cell proliferation and contributed to altered follicle formation, typical of PCOS [ 69 ]. Table 4 summarizes the main studies about CFTR and reproductive system disorders.
CFTR and cardiovascular system
CFTR seems to be involved in blood pressure regulation. CFTR knock-out mice developed higher mean blood pressure compared with controls ( p < 0.05). This evidence was supported by CFTR downregulation in a model of induced hypertension, where a high fructose and salt diet reduced With-No-Lysine K (WNK) kinase expression in arteries and subsequently CFTR expression, leading to increased vascular constriction. This was further supported by the evidence that CFTR knock-out mice fed with high fructose and salt dose did not show increased mean blood pressure ( p < 0.05). [ 70 ]. However, further studies are needed to confirm these preliminary data.
Mutations in CFTR gene are often associated with CF. In this review, we highlighted the broad spectrum of alterations reported for this gene, which may be involved in the pathogenesis of other diseases [Fig. 1 ]. The extensive application of genetic testing provides evidence to link the CFTR gene mutation with different conditions and characterize their genotype-phenotypes. The importance of these new insights in the role of CFTR relies on the possibility of making it a novel therapeutic target. In this context, results obtained by the new CFTR modulators in CF are promising, highlighting their ability to modify the disease course. Therefore, they might improve treatment of high burden diseases through precision medicine [ 71 ]. However, despite the ubiquitous role of CFTR is various organ systems, there is still a dearth of CFTR-based therapeutics. Some of the reasons might be unstable pharmacokinetics, cross-talk with other cellular proteins, and sampling issues for clinical trials. These points have to be considered while developing CFTR-based therapeutic interventions.

Summary of main functions of the CFTR
Availability of data and material
Not applicable.
Code availability
Abbreviations.
Protein kinase B
Adenosine triphosphate
Cyclic adenosine monophosphate
Congenital bilateral absence of the vas deferens
- Celiac disease
Cystic fibrosis
Cystic fibrosis transmembrane conductance regulator
Cyclic guanosine monophosphate
Chronic obstructive pulmonary disease
cAMP response element-binding protein
Chymotrypsinogen C
Extracellular-regulated kinase
Forced expiratory volume in 1 s
Follicle-stimulating hormone
Heat shock protein 90
Idiopathic chronic pancreatitis
Janus kinase 2
Reduced micro-RNA
Matrix metalloproteinase 11
Nucleotide-binding domain-1
NF-κB inhibitor alpha
Nuclear factor kappa-light-chain-enhancer of activated B cells
Polycystic ovarian syndrome
Protein phosphatase 2A
Cationic trypsinogen gene
Primary sclerosing cholangitis
Serine peptidase inhibitor
Signal transducer and activator of transcription-3
Tissue transglutaminase 2
Urokinase plasminogen activator
Vascular endothelial growth factor
Vasoactive intestinal peptide
With-no-lysine K
Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, Grzelczak Z, Zielenski J, Lok S, Plavsic N, Chou JL, et al (1989) Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science 245(4922):1066-73. https://doi.org/10.1126/science.2475911 . Erratum in: Science 1989 Sep 29;245(4925):1437
http://www.genet.sickkids.on.ca/ Accessed on 21th January, 2021
Parisi GF, Cutello S, Di Dio G, Rotolo N, La Rosa M, Leonardi S (2013) Phenotypic expression of the p.Leu1077Pro CFTR mutation in Sicilian cystic fibrosis patients. BMC Res Notes 6:461. https://doi.org/10.1186/1756-0500-6-461
Article CAS PubMed PubMed Central Google Scholar
Leonardi S, Parisi GF, Capizzi A, Manti S, Cuppari C, Scuderi MG, Rotolo N, Lanzafame A, Musumeci M, Salpietro C (2016) YKL-40 as marker of severe lung disease in cystic fibrosis patients. J Cyst Fibros 15(5):583–586. https://doi.org/10.1016/j.jcf.2015.12.020
Article CAS PubMed Google Scholar
Spicuzza L, Parisi GF, Tardino L, Ciancio N, Nenna R, Midulla F, Leonardi S (2018) Exhaled markers of antioxidant activity and oxidative stress in stable cystic fibrosis patients with moderate lung disease. J Breath Res 12(2):026010. https://doi.org/10.1088/1752-7163/aa9b39
Parisi GF, Portale A, Papale M, Tardino L, Rotolo N, Licari A, Leonardi S (2019) Successful treatment with omalizumab of allergic bronchopulmonary aspergillosis in patients with cystic fibrosis: case reports and literature review. J Allergy Clin Immunol Pract 7(5):1636–1638. https://doi.org/10.1016/j.jaip.2019.01.056
Article PubMed Google Scholar
Bombieri C, Claustres M, De Boeck K, Derichs N, Dodge J, Girodon E, Sermet I, Schwarz M, Tzetis M, Wilschanski M, Bareil C, Bilton D, Castellani C, Cuppens H, Cutting GR, Drevínek P, Farrell P, Elborn JS, Jarvi K, Kerem B, Kerem E, Knowles M, Macek M Jr, Munck A, Radojkovic D, Seia M, Sheppard DN, Southern KW, Stuhrmann M, Tullis E, Zielenski J, Pignatti PF, Ferec C (2011) Recommendations for the classification of diseases as CFTR-related disorders. J Cyst Fibros 10(Suppl 2):S86-102. https://doi.org/10.1016/S1569-1993(11)60014-3
Galluzzi L, Kroemer G (2019) Etiological involvement of CFTR in apparently unrelated human diseases. Mol Cell Oncol. https://doi.org/10.1080/23723556.2018.1558874
Zhang J, Wang Y, Jiang X, Chan HC (2018) Cystic fibrosis transmembrane conductance regulator-emerging regulator of cancer. Cell Mol Life Sci 75(10):1737–1756. https://doi.org/10.1007/s00018-018-2755-6
Shi Z, Wei J, Na R, Resurreccion WK, Zheng SL, Hulick PJ, Helfand BT, Talamonti MS, Xu J (2021) Cystic fibrosis F508del carriers and cancer risk: Results from the UK Biobank. Int J Cancer 148(7):1658–1664. https://doi.org/10.1002/ijc.33431
Çolak Y, Nordestgaard BG, Afzal S (2020) Morbidity and mortality in carriers of the cystic fibrosis mutation CFTR Phe508del in the general population. Eur Respir J. https://doi.org/10.1183/13993003.00558-2020
Schubert S, Traub F, Brakensiek K, von Kopylow K, Marohn B, Maelzer M, Gaedcke J, Kreipe H, Stuhrmann M (2014) CFTR, SPINK1, PRSS1, and CTRC mutations are not associated with pancreatic cancer in German patients. Pancreas 43(7):1078–1082. https://doi.org/10.1097/MPA.0000000000000166
Cazacu IM, Farkas N, Garami A, Balaskó M, Mosdósi B, Alizadeh H, Gyöngyi Z, Rakonczay Z Jr, Vigh É, Habon T, Czopf L, Lazarescu MA, Erőss B, Sahin-Tóth M, Hegyi P (2018) Pancreatitis-associated genes and pancreatic cancer risk: a systematic review and meta-analysis. Pancreas 47(9):1078–1086. https://doi.org/10.1097/MPA.0000000000001145
Esteller M (2008) Epigenetics in cancer. N Engl J Med. https://doi.org/10.1056/nejmra072067
Li W, Wang C, Peng X, Zhang H, Huang H, Liu H (2018) CFTR inhibits the invasion and growth of esophageal cancer cells by inhibiting the expression of NF-κB. Cell Biol Int 42(12):1680–1687. https://doi.org/10.1002/cbin.11069
Liu C, Song C, Li J, Sun Q (2020) CFTR Functions as a tumor suppressor and is regulated by DNA methylation in colorectal cancer. Cancer Manag Res 12:4261–4270. https://doi.org/10.2147/CMAR.S248539
Than BL, Linnekamp JF, Starr TK, Largaespada DA, Rod A, Zhang Y, Bruner V, Abrahante J, Schumann A, Luczak T, Walter J, Niemczyk A, O'Sullivan MG, Medema JP, Fijneman RJ, Meijer GA, Van den Broek E, Hodges CA, Scott PM, Vermeulen L, Cormier RT. (2016) CFTR is a tumor suppressor gene in murine and human intestinal cancer. Oncogene 35 (32):4179-87. https://doi.org/10.1038/onc.2015.483 Erratum in: Oncogene. 2017, 36(24):3504
Liu K, Jin H, Guo Y, Liu Y, Wan Y, Zhao P, Zhou Z, Wang J, Wang M, Zou C, Wu W, Cheng Z, Dai Y (2019) CFTR interacts with Hsp90 and regulates the phosphorylation of AKT and ERK1/2 in colorectal cancer cells. FEBS Open Bio. 9(6):1119–1127. https://doi.org/10.1002/2211-5463.12641
Shin Y, Kim M, Won J, Kim J, Oh SB, Lee JH, Park K (2020) Epigenetic modification of CFTR in head and neck cancer. J Clin Med 9(3):734. https://doi.org/10.3390/jcm9030734
Article CAS PubMed Central Google Scholar
Liu K, Dong F, Gao H, Guo Y, Li H, Yang F, Zhao P, Dai Y, Wang J, Zhou W, Zou C (2020) Promoter hypermethylation of the CFTR gene as a novel diagnostic and prognostic marker of breast cancer. Cell Biol Int 44(2):603–609. https://doi.org/10.1002/cbin.11260
Son JW, Kim YJ, Cho HM, Lee SY, Lee SM, Kang JK, Lee JU, Lee YM, Kwon SJ, Choi E, Na MJ, Park JY, Kim DS (2011) Promoter hypermethylation of the CFTR gene and clinical/pathological features associated with non-small cell lung cancer. Respirology 16(8):1203–1209. https://doi.org/10.1111/j.1440-1843.2011.01994.x
Yu J, Zhu T, Wang Z, Zhang H, Qian Z, Xu H, Gao B, Wang W, Gu L, Meng J, Wang J, Feng X, Li Y, Yao X, Zhu J (2007) A novel set of DNA methylation markers in urine sediments for sensitive/specific detection of bladder cancer. Clin Cancer Res 13(24):7296–7304. https://doi.org/10.1158/1078-0432.CCR-07-0861
Moribe T, Iizuka N, Miura T, Kimura N, Tamatsukuri S, Ishitsuka H, Hamamoto Y, Sakamoto K, Tamesa T, Oka M (2009) Methylation of multiple genes as molecular markers for diagnosis of a small, well-differentiated hepatocellular carcinoma. Int J Cancer 125(2):388–397. https://doi.org/10.1002/ijc.24394
Zhang JT, Jiang XH, Xie C, Cheng H, Da Dong J, Wang Y, Fok KL, Zhang XH, Sun TT, Tsang LL, Chen H, Sun XJ, Chung YW, Cai ZM, Jiang WG, Chan HC (2013) Downregulation of CFTR promotes epithelial-to-mesenchymal transition and is associated with poor prognosis of breast cancer. Biochim Biophys Acta 1833(12):2961–2969. https://doi.org/10.1016/j.bbamcr.2013.07.021
Tizzano EF, Silver MM, Chitayat D, Benichou JC, Buchwald M (1994) Differential cellular expression of cystic fibrosis transmembrane regulator in human reproductive tissues: clues for the infertility in patients with cystic fibrosis. Am J Pathol 144(5):906
CAS PubMed PubMed Central Google Scholar
Xu J, Yong M, Li J, Dong X, Yu T, Fu X, Hu L (2015) High level of CFTR expression is associated with tumor aggression and knockdown of CFTR suppresses proliferation of ovarian cancer in vitro and in vivo. Oncol Rep 33(5):2227–2234. https://doi.org/10.3892/or.2015.3829
Peng X, Wu Z, Yu L, Li J, Xu W, Chan HC, Zhang Y, Hu L (2012) Overexpression of cystic fibrosis transmembrane conductance regulator (CFTR) is associated with human cervical cancer malignancy, progression and prognosis. Gynecol Oncol 125(2):470–476. https://doi.org/10.1016/j.ygyno.2012.02.015
Wu Z, Li J, Zhang Y, Hu L, Peng X (2020) CFTR regulates the proliferation, migration and invasion of cervical cancer cells by inhibiting the NF-κB signalling pathway. Cancer Manag Res. https://doi.org/10.2147/CMAR.S252296
Article PubMed PubMed Central Google Scholar
Xia X, Wang J, Liu Y, Yue M (2017) Lower cystic fibrosis transmembrane conductance regulator (CFTR) promotes the proliferation and migration of endometrial carcinoma. Med Sci Monit. https://doi.org/10.12659/MSM.899341
Xie C, Jiang XH, Zhang JT, Sun TT, Dong JD, Sanders AJ, Diao RY, Wang Y, Fok KL, Tsang LL, Yu MK, Zhang XH, Chung YW, Ye L, Zhao MY, Guo JH, Xiao ZJ, Lan HY, Ng CF, Lau KM, Cai ZM, Jiang WG, Chan HC (2013) CFTR suppresses tumor progression through miR-193b targeting urokinase plasminogen activator (uPA) in prostate cancer. Oncogene 32(18):2282-91-2291.e1–7. https://doi.org/10.1038/onc.2012.251
Article CAS Google Scholar
Tu Z, Chen Q, Zhang JT, Jiang X, Xia Y, Chan HC (2016) CFTR is a potential marker for nasopharyngeal carcinoma prognosis and metastasis. Oncotarget. https://doi.org/10.18632/oncotarget.12762
Zhong X, Chen HQ, Yang XL, Wang Q, Chen W, Li C (2019) CFTR activation suppresses glioblastoma cell proliferation, migration and invasion. Biochem Biophys Res Commun. https://doi.org/10.1016/j.bbrc.2018.12.080
Zhu Q, Li H, Liu Y, Jiang L (2017) Knockdown of CFTR enhances sensitivity of prostate cancer cells to cisplatin via inhibition of autophagy. Neoplasma. https://doi.org/10.4149/neo_2017_508
Li H, Ma N, Wang J, Wang Y, Yuan C, Wu J, Luo M, Yang J, Chen J, Shi J, Liu X (2018) Nicotine induces progressive properties of lung adenocarcinoma A549 cells by inhibiting cystic fibrosis transmembrane conductance regulator (CFTR) expression and plasma membrane localization. Technol Cancer Res Treat 1(17):1533033818809984. https://doi.org/10.1177/1533033818809984
Yang X, Yan T, Gong Y, Liu X, Sun H, Xu W, Wang C, Naren D, Zheng Y (2017) High CFTR expression in Philadelphia chromosome-positive acute leukemia protects and maintains continuous activation of BCR-ABL and related signaling pathways in combination with PP2A. Oncotarget 8(15):24437–24448. https://doi.org/10.18632/oncotarget.15510
Liu M, Liao H, Chen Y, Lin Z, Liu Y, Zhang X, Chan HC, Sun H (2019) Treatment of human T-cell acute lymphoblastic leukemia cells with CFTR inhibitor CFTRinh-172. Leuk Res 86:106225. https://doi.org/10.1016/j.leukres.2019.106225
Parisi GF, Papale M, Rotolo N, Aloisio D, Tardino L, Scuderi MG, Di Benedetto V, Nenna R, Midulla F, Leonardi S (2017) Severe disease in cystic fibrosis and fecal calprotectin levels. Immunobiology 222(3):582–586. https://doi.org/10.1016/j.imbio.2016.11.005
Cohn JA, Neoptolemos JP, Feng J, Yan J, Jiang Z, Greenhalf W, McFaul C, Mountford R, Sommer SS (2005) Increased risk of idiopathic chronic pancreatitis in cystic fibrosis carriers. Hum Mutat 26(4):303–307. https://doi.org/10.1002/humu.20232
Schneider A, Larusch J, Sun X, Aloe A, Lamb J, Hawes R, Cotton P, Brand RE, Anderson MA, Money ME, Banks PA, Lewis MD, Baillie J, Sherman S, Disario J, Burton FR, Gardner TB, Amann ST, Gelrud A, George R, Rockacy MJ, Kassabian S, Martinson J, Slivka A, Yadav D, Oruc N, Barmada MM, Frizzell R, Whitcomb DC (2011) Combined bicarbonate conductance-impairing variants in CFTR and SPINK1 variants are associated with chronic pancreatitis in patients without cystic fibrosis. Gastroenterologyb 140(1):162–71. https://doi.org/10.1053/j.gastro.2010.10.045
Rosendahl J, Landt O, Bernadova J, Kovacs P, Teich N, Bödeker H, Keim V, Ruffert C, Mössner J, Kage A, Stumvoll M, Groneberg D, Krüger R, Luck W, Treiber M, Becker M, Witt H (2013) CFTR, SPINK1, CTRC and PRSS1 variants in chronic pancreatitis: is the role of mutated CFTR overestimated? Gut 62(4):582–592. https://doi.org/10.1136/gutjnl-2011-300645
Raphael KL, Willingham FF (2016) Hereditary pancreatitis: current perspectives. Clin Exp Gastroenterol 9:197–207. https://doi.org/10.2147/CEG.S84358
Zou WB, Tang XY, Zhou DZ, Qian YY, Hu LH, Yu FF, Yu D, Wu H, Deng SJ, Lin JH, Zhao AJ, Zhao ZH, Wu HY, Zhu JH, Qian W, Wang L, Xin L, Wang MJ, Wang LJ, Fang X, He L, Masson E, Cooper DN, Férec C, Li ZS, Chen JM, Liao Z (2018) SPINK1, PRSS1, CTRC, and CFTR genotypes influence disease onset and clinical outcomes in chronic pancreatitis. Clin Transl Gastroenterol 9(11):204. https://doi.org/10.1038/s41424-018-0069-5
de Jonge HR, Ardelean MC, Bijvelds MJC, Vergani P (2020) Strategies for cystic fibrosis transmembrane conductance regulator inhibition: from molecular mechanisms to treatment for secretory diarrhoeas. FEBS Lett 594(23):4085–4108. https://doi.org/10.1002/1873-3468.13971
Debray D, Rainteau D, Barbu V, Rouahi M, El Mourabit H, Lerondel S, Rey C, Humbert L, Wendum D, Cottart CH, Dawson P, Chignard N, Housset C (2012) Defects in gallbladder emptying and bile acid homeostasis in mice with cystic fibrosis transmembrane conductance regulator deficiencies. Gastroenterology 142(7):1581–91.e6. https://doi.org/10.1053/j.gastro.2012.02.033
Lazaridis KN, LaRusso NF (2016) Primary sclerosing cholangitis. N Engl J Med 375(12):1161–1170. https://doi.org/10.1056/NEJMra1506330
Eaton JE, Talwalkar JA, Lazaridis KN, Gores GJ, Lindor KD (2013) Pathogenesis of primary sclerosing cholangitis and advances in diagnosis and management. Gastroenterology. https://doi.org/10.1053/j.gastro.2013.06.052
Sheth S, Shea JC, Bishop MD, Chopra S, Regan MM, Malmberg E, Walker C, Ricci R, Tsui LC, Durie PR, Zielenski J, Freedman SD (2003) Increased prevalence of CFTR mutations and variants and decreased chloride secretion in primary sclerosing cholangitis. Hum Genet 113(3):286–292. https://doi.org/10.1007/s00439-003-0963-z
Werlin S, Scotet V, Uguen K, Audrezet MP, Cohen M, Yaakov Y, Safadi R, Ilan Y, Konikoff F, Galun E, Mizrahi M, Slae M, Sayag S, Cohen-Cymberknoh M, Wilschanski M, Ferec C (2018) Primary sclerosing cholangitis is associated with abnormalities in CFTR. J Cyst Fibros 17(5):666–671. https://doi.org/10.1016/j.jcf.2018.04.005
Gallegos-Orozco JF, Yurk EC, Wang N, Rakela J, Charlton MR, Cutting GR, Balan V (2005) Lack of association of common cystic fibrosis transmembrane conductance regulator gene mutations with primary sclerosing cholangitis. Am J Gastroenterol. 100(4):874–8. https://doi.org/10.1111/j.1572-0241.2005.41072.x
Lionetti E, Castellaneta S, Francavilla R, Pulvirenti A, Tonutti E, Amarri S, Barbato M, Barbera C, Barera G, Bellantoni A, Castellano E, Guariso G, Limongelli MG, Pellegrino S, Polloni C, Ughi C, Zuin G, Fasano A, Catassi C (2014) SIGENP (Italian society of pediatric gastroenterology, hepatology, and nutrition) working group on weaning and CD risk introduction of gluten, HLA status, and the risk of celiac disease in children. N Engl J Med 371(14):1295–303. https://doi.org/10.1056/NEJMoa1400697
Walkowiak J, Blask-Osipa A, Lisowska A, Oralewska B, Pogorzelski A, Cichy W, Sapiejka E, Kowalska M, Korzon M, Szaflarska-Popławska A (2010) Cystic fibrosis is a risk factor for celiac disease. Acta Biochim Pol 57(1):115–118
Lionetti E, Gatti S, Pulvirenti A, Catassi C (2015) Celiac disease from a global perspective. Best Pract Res Clin Gastroenterol 29(3):365–379. https://doi.org/10.1016/j.bpg.2015.05.004
Villella VR, Venerando A, Cozza G, Esposito S, Ferrari E, Monzani R, Spinella MC, Oikonomou V, Renga G, Tosco A, Rossin F, Guido S, Silano M, Garaci E, Chao YK, Grimm C, Luciani A, Romani L, Piacentini M, Raia V, Kroemer G, Maiuri L (2019) A pathogenic role for cystic fibrosis transmembrane conductance regulator in celiac disease. EMBO J 38(2):100101. https://doi.org/10.15252/embj.2018100101
Luciani A, Villella VR, Esposito S, Brunetti-Pierri N, Medina D, Settembre C, Gavina M, Pulze L, Giardino I, Pettoello-Mantovani M, D’Apolito M, Guido S, Masliah E, Spencer B, Quaratino S, Raia V, Ballabio A, Maiuri L (2010) Defective CFTR induces aggresome formation and lung inflammation in cystic fibrosis through ROS-mediated autophagy inhibition. Nat Cell Biol 12(9):863–875. https://doi.org/10.1038/ncb2090
Maiuri L, Villella VR, Piacentini M, Raia V, Kroemer G (2019) Defective proteostasis in celiac disease as a new therapeutic target. Cell Death Dis. https://doi.org/10.1038/s41419-019-1392-9
Esposito S, Villella VR, Ferrari E, Monzani R, Tosco A, Rossin F, D’Eletto M, Castaldo A, Luciani A, Silano M, Bona G, Marseglia GL, Romani L, Piacentini M, Raia V, Kroemer G, Maiuri L (2019) Genistein antagonizes gliadin-induced CFTR malfunction in models of celiac disease. Aging (Albany NY) 11(7):2003–2019. https://doi.org/10.18632/aging.101888
Zeng M, Szymczak M, Ahuja M, Zheng C, Yin H, Swaim W, Chiorini JA, Bridges RJ, Muallem S (2017) Restoration of CFTR activity in ducts rescues acinar cell function and reduces inflammation in pancreatic and salivary glands of mice. Gastroenterology 153(4):1148–1159. https://doi.org/10.1053/j.gastro.2017.06.011
Fernandez Fernandez E, De Santi C, De Rose V, Greene CM (2018) CFTR dysfunction in cystic fibrosis and chronic obstructive pulmonary disease. Expert Rev Respir Med 12(6):483–492. https://doi.org/10.1080/17476348.2018
Raju SV, Jackson PL, Courville CA, McNicholas CM, Sloane PA, Sabbatini G, Tidwell S, Tang LP, Liu B, Fortenberry JA, Jones CW, Boydston JA, Clancy JP, Bowen LE, Accurso FJ, Blalock JE, Dransfield MT, Rowe SM (2013) Cigarette smoke induces systemic defects in cystic fibrosis transmembrane conductance regulator function. Am J Respir Crit Care Med 188(11):1321–1330. https://doi.org/10.1164/rccm.201304-0733OC
Rennolds J, Butler S, Maloney K, Boyaka PN, Davis IC, Knoell DL, Parinandi NL, Cormet-Boyaka E (2010) Cadmium regulates the expression of the CFTR chloride channel in human airway epithelial cells. Toxicol Sci 116(1):349–58. https://doi.org/10.1093/toxsci/kfq101
Bomberger JM, Coutermarsh BA, Barnaby RL, Stanton BA (2012) Arsenic promotes ubiquitinylation and lysosomal degradation of cystic fibrosis transmembrane conductance regulator (CFTR) chloride channels in human airway epithelial cells. J Biol Chem. https://doi.org/10.1074/jbc.M111.338855
Rowe SM, Jones I, Dransfield MT, Haque N, Gleason S, Hayes KA, Kulmatycki K, Yates DP, Danahay H, Gosling M, Rowlands DJ, Grant SS (2020) Efficacy and safety of the CFTR potentiator icenticaftor (QBW251) in COPD: results from a phase 2 randomized trial. Int J Chron Obstruct Pulmon Dis 5(15):2399–2409. https://doi.org/10.2147/COPD.S257474
Article Google Scholar
Licari A, Manti S, Castagnoli R, Parisi GF, Salpietro C, Leonardi S, Marseglia GL (2019) Targeted therapy for severe asthma in children and adolescents: current and future perspectives. Paediatr Drugs 21(4):215–237. https://doi.org/10.1007/s40272-019-00345-7
Leonardi S, Filippelli M, Lanzafame A, Parisi G, Mistrello G, Musumeci M, Torrisi V, Musumeci S, Cuppari C (2019) Serum YKL-40 in children with asthma. J Biol Regul Homeost Agents 29(21):114–9
Google Scholar
Nielsen AO, Qayum S, Bouchelouche PN, Laursen LC, Dahl R, Dahl M (2016) Risk of asthma in heterozygous carriers for cystic fibrosis: a meta-analysis. J Cyst Fibros. https://doi.org/10.1016/j.jcf.2016.06.001
Yu J, Chen Z, Ni Y, Li Z (2012) CFTR mutations in men with congenital bilateral absence of the vas deferens (CBAVD): a systemic review and meta-analysis. Hum Reprod 27(1):25–35. https://doi.org/10.1093/humrep/der377
Ma C, Wang R, Li T, Li H, Wang B (2020) Analysis of CNVs of CFTR gene in Chinese Han population with CBAVD. Mol Genet Genomic Med. https://doi.org/10.1002/mgg3.1506
Chen H, Guo JH, Lu YC, Ding GL, Yu MK, Tsang LL, Fok KL, Liu XM, Zhang XH, Chung YW, Huang P, Huang H, Chan HC (2012) Impaired CFTR-dependent amplification of FSH-stimulated estrogen production in cystic fibrosis and PCOS. J Clin Endocrinol Metab 97(3):923–932. https://doi.org/10.1210/jc.2011-1363
Chen H, Guo JH, Zhang XH, Chan HC (2015) Defective CFTR-regulated granulosa cell proliferation in polycystic ovarian syndrome. Reproduction. https://doi.org/10.1530/REP-14-0368
Zhang Y-P, Ye LL, Yuan H, Duan DD (2020) CFTR plays an important role in the regulation of vascular resistance and high-fructose/salt-diet induced hypertension in mice. J Cyst Fibros Off J Eur Cyst Fibros Soc. https://doi.org/10.1016/j.jcf.2020.11.014
Dagenais RVE, Su VCH, Quon BS (2020) Real-world safety of CFTR modulators in the treatment of cystic fibrosis: a systematic review. J Clin Med 10(1):23. https://doi.org/10.3390/jcm10010023
Download references
Acknowledgements
Not applicable
The authors did not receive any funding for the research.
Author information
Authors and affiliations.
Department of Clinical and Experimental Medicine, University of Catania, Via Santa Sofia, 78, 95123, Catania, Italy
Giuseppe Fabio Parisi, Federico Mòllica, Alessandro Giallongo, Maria Papale, Sara Manti & Salvatore Leonardi
You can also search for this author in PubMed Google Scholar
Contributions
GFP, FM, and AG wrote the manuscript; MP and SM performed the research and reviewed the manuscript; SL supervised the work; all authors have read and approve the final manuscript.
Corresponding author
Correspondence to Giuseppe Fabio Parisi .
Ethics declarations
Ethical approval and consent to participate, consent for publication, competing interests.
The authors have no conflicts of interest to disclose that could be perceived as prejudicing the impartiality of the research reported.
Additional information
Publisher's note.
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/ .
Reprints and permissions
About this article
Cite this article.
Parisi, G.F., Mòllica, F., Giallongo, A. et al. Cystic fibrosis transmembrane conductance regulator (CFTR): beyond cystic fibrosis. Egypt J Med Hum Genet 23 , 94 (2022). https://doi.org/10.1186/s43042-022-00308-7
Download citation
Received : 30 August 2021
Accepted : 28 April 2022
Published : 16 May 2022
DOI : https://doi.org/10.1186/s43042-022-00308-7
Share this article
Anyone you share the following link with will be able to read this content:
Sorry, a shareable link is not currently available for this article.
Provided by the Springer Nature SharedIt content-sharing initiative
- Gastrointestinal disorders
- Autoimmune disease
Information
- Author Services
Initiatives
You are accessing a machine-readable page. In order to be human-readable, please install an RSS reader.
All articles published by MDPI are made immediately available worldwide under an open access license. No special permission is required to reuse all or part of the article published by MDPI, including figures and tables. For articles published under an open access Creative Common CC BY license, any part of the article may be reused without permission provided that the original article is clearly cited. For more information, please refer to https://www.mdpi.com/openaccess .
Feature papers represent the most advanced research with significant potential for high impact in the field. A Feature Paper should be a substantial original Article that involves several techniques or approaches, provides an outlook for future research directions and describes possible research applications.
Feature papers are submitted upon individual invitation or recommendation by the scientific editors and must receive positive feedback from the reviewers.
Editor’s Choice articles are based on recommendations by the scientific editors of MDPI journals from around the world. Editors select a small number of articles recently published in the journal that they believe will be particularly interesting to readers, or important in the respective research area. The aim is to provide a snapshot of some of the most exciting work published in the various research areas of the journal.
Original Submission Date Received: .
- Active Journals
- Find a Journal
- Proceedings Series
- For Authors
- For Reviewers
- For Editors
- For Librarians
- For Publishers
- For Societies
- For Conference Organizers
- Open Access Policy
- Institutional Open Access Program
- Special Issues Guidelines
- Editorial Process
- Research and Publication Ethics
- Article Processing Charges
- Testimonials
- Preprints.org
- SciProfiles
- Encyclopedia
Article Menu

- Subscribe SciFeed
- Recommended Articles
- Author Biographies
- PubMed/Medline
- Google Scholar
- on Google Scholar
- Table of Contents
Find support for a specific problem in the support section of our website.
Please let us know what you think of our products and services.
Visit our dedicated information section to learn more about MDPI.
JSmol Viewer
Functional consequences of cftr interactions in cystic fibrosis, 1. introduction, 1.1. from discovery and diagnostics to therapy in cystic fibrosis, 1.2. history of cystic fibrosis, 1.3. multi-organ disease manifestations in cf, 1.3.1. cf lung, 1.3.2. cf pancreas, 1.3.3. cf gut, 1.4. cystic fibrosis transmembrane conductance regulator (cftr) gene, 1.5. cystic fibrosis transmembrane conductance regulator (cftr) protein, 1.6. molecular structure of cftr ion channel, 1.7. addressing the cftr protein defect using small molecules-cftr modulators, 1.7.1. cf modulators in clinical application, 1.7.2. other cftr modulators for potential clinical application, 1.8. non-cftr therapies in cf, 1.9. prospective gene modification therapies in cf, 1.10. cftr expression defined cellular compositions in the lung, 1.10.1. ionocytes, 1.10.2. secretory cells, 1.10.3. basal cells, 1.10.4. ciliated cells, 1.11. altered protein interactions determine the etiological course of cftr-related disorders, 1.11.1. snapshot of the molecular communication between cftr ppis and cf by various proteomic approaches, 1.11.2. cellular sub-compartmental state and composition of cftr ppis and ppi enabled highly compartmentalized cftr signalosomes, author contributions, acknowledgments, conflicts of interest.
- Davis, P.B. Cystic fibrosis since 1938. Am. J. Respir. Crit. Care Med. 2006 , 173 , 475–482. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Kerem, B.-S.; Rommens, J.M.; Buchanan, J.A.; Markiewicz, D.; Cox, T.K.; Chakravarti, A.; Buchwald, M.; Tsui, L.-C. Identification of the cystic fibrosis gene: Genetic analysis. Science 1989 , 245 , 1073–1080. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Riordan, J.R.; Rommens, J.M.; Kerem, B.-s.; Alon, N.; Rozmahel, R.; Grzelczak, Z.; Zielenski, J.; Lok, S.; Plavsic, N.; Chou, J.-L. Identification of the cystic fibrosis gene: Cloning and characterization of complementary DNA. Science 1989 , 245 , 1066–1073. [ Google Scholar ] [ CrossRef ]
- Davis, S.D.; Rosenfeld, M.; Chmiel, J. Cystic Fibrosis: A Multi-Organ System Approach ; Springer Nature: New York, NY, USA, 2020. [ Google Scholar ]
- Andersen, D.H. Cystic fibrosis of the pancreas and its relation to celiac disease: A clinical and pathologic study. Am. J. Dis. Child. 1938 , 56 , 344–399. [ Google Scholar ] [ CrossRef ]
- Farber, S. Pancreatic function and disease in early life. V. Pathologic changes associated with pancreatic insufficiency in early life. Arch. Pathol. 1944 , 37 , 238–250. [ Google Scholar ]
- di Sant’Agnese, P.A.; Darling, R.C.; Perera, G.A.; Shea, E. Abnormal electrolyte composition of sweat in cystic fibrosis of the pancreas: Clinical significance and relationship to the disease. Pediatrics 1953 , 12 , 549–563. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Gibson, L.E.; Cooke, R.E. A test for concentration of electrolytes in sweat in cystic fibrosis of the pancreas utilizing pilocarpine by iontophoresis. Pediatrics 1959 , 23 , 545–549. [ Google Scholar ] [ CrossRef ]
- Matthews, L.W.; Doershuk, C.F.; Wise, M.; Eddy, G.; Nudelman, H.; Spector, S. A therapeutic regimen for patients with cystic fibrosis. J. Pediatr. 1964 , 65 , 558–575. [ Google Scholar ] [ CrossRef ]
- Anderson, M.P.; Rich, D.P.; Gregory, R.J.; Smith, A.E.; Welsh, M.J. Generation of cAMP-activated chloride currents by expression of CFTR. Science 1991 , 251 , 679–682. [ Google Scholar ] [ CrossRef ]
- Denning, G.M.; Anderson, M.P.; Amara, J.F.; Marshall, J.; Smith, A.E.; Welsh, M.J. Processing of mutant cystic fibrosis transmem-brane conductance regulator is temperature-sensitive. Nature 1992 , 358 , 761–764. [ Google Scholar ] [ CrossRef ]
- Liu, F.; Zhang, Z.; Csanády, L.; Gadsby, D.C.; Chen, J. Molecular structure of the human CFTR ion channel. Cell 2017 , 169 , 85–95.e88. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Lewis, H.A.; Buchanan, S.G.; Burley, S.K.; Conners, K.; Dickey, M.; Dorwart, M.; Fowler, R.; Gao, X.; Guggino, W.B.; Hendrickson, W.A.; et al. Structure of nucleotide-binding domain 1 of the cystic fibrosis transmembrane conductance regulator. EMBO J. 2004 , 23 , 282–293. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Li, C.; Naren, A.P. CFTR chloride channel in the apical compartments: Spatiotemporal coupling to its interacting partners. Integr. Biol. 2010 , 2 , 161–177. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Chen, Q.; Shen, Y.; Zheng, J. A review of cystic fibrosis: Basic and clinical aspects. Anim. Model. Exp. Med. 2021 , 4 , 220–232. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Wang, S.; Li, M. Molecular studies of CFTR interacting proteins. Pflug. Arch. 2001 , 443 (Suppl. S1), S62–S64. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Heuser, T.; Raytchev, M.; Krell, J.; Porter, M.E.; Nicastro, D. The dynein regulatory complex is the nexin link and a major regulatory node in cilia and flagella. J. Cell Biol. 2009 , 187 , 921–933. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Brooks, E.R.; Wallingford, J.B. Multiciliated cells. Curr. Biol. 2014 , 24 , R973–R982. [ Google Scholar ] [ CrossRef ]
- Bustamante-Marin, X.M.; Ostrowski, L.E. Cilia and mucociliary clearance. Cold Spring Harb. Perspect. Biol. 2017 , 9 , a028241. [ Google Scholar ] [ CrossRef ]
- Whitsett, J.A. Airway epithelial differentiation and mucociliary clearance. Ann. Am. Thorac. Soc. 2018 , 15 (Suppl. S3), S143–S148. [ Google Scholar ] [ CrossRef ]
- Houtmeyers, E.; Gosselink, R.; Gayan-Ramirez, G.; Decramer, M. Regulation of mucociliary clearance in health and disease. Eur. Respir. J. 1999 , 13 , 1177–1188. [ Google Scholar ] [ CrossRef ]
- Zabner, J.; Smith, J.J.; Karp, P.H.; Widdicombe, J.H.; Welsh, M.J. Loss of CFTR Chloride Channels Alters Salt Absorption by Cystic Fibrosis Airway Epithelia In Vitro. Mol. Cell 1998 , 2 , 397–403. [ Google Scholar ] [ CrossRef ]
- Coakley, R.D.; Grubb, B.R.; Paradiso, A.M.; Gatzy, J.T.; Johnson, L.G.; Kreda, S.M.; O’Neal, W.K.; Boucher, R.C. Abnormal surface liquid pH regulation by cultured cystic fibrosis bronchial epithelium. Proc. Natl. Acad. Sci. USA 2003 , 100 , 16083–16088. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Song, Y.; Salinas, D.; Nielson, D.W.; Verkman, A. Hyperacidity of secreted fluid from submucosal glands in early cystic fibrosis. Am. J. Physiol. Cell Physiol. 2006 , 290 , C741–C749. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Tate, S.; MacGregor, G.; Davis, M.; Innes, J.; Greening, A. Airways in cystic fibrosis are acidified: Detection by exhaled breath condensate. Thorax 2002 , 57 , 926–929. [ Google Scholar ] [ CrossRef ]
- Pezzulo, A.A.; Tang, X.X.; Hoegger, M.J.; Abou Alaiwa, M.H.; Ramachandran, S.; Moninger, T.O.; Karp, P.H.; Wohlford-Lenane, C.L.; Haagsman, H.P.; Van Eijk, M. Reduced airway surface pH impairs bacterial killing in the porcine cystic fibrosis lung. Nature 2012 , 487 , 109–113. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- McShane, D.; Davies, J.; Davies, M.; Bush, A.; Geddes, D.; Alton, E. Airway surface pH in subjects with cystic fibrosis. Eur. Respir. J. 2003 , 21 , 37–42. [ Google Scholar ] [ CrossRef ]
- Abou Alaiwa, M.H.; Beer, A.M.; Pezzulo, A.A.; Launspach, J.L.; Horan, R.A.; Stoltz, D.A.; Starner, T.D.; Welsh, M.J.; Zabner, J. Neonates with cystic fibrosis have a reduced nasal liquid pH; a small pilot study. J. Cyst. Fibros. 2014 , 13 , 373–377. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Boucher, R.C. Muco-obstructive lung diseases. N. Engl. J. Med. 2019 , 380 , 1941–1953. [ Google Scholar ] [ CrossRef ]
- Tilley, A.E.; Walters, M.S.; Shaykhiev, R.; Crystal, R.G. Cilia dysfunction in lung disease. Annu. Rev. Physiol. 2015 , 77 , 379–406. [ Google Scholar ] [ CrossRef ]
- Sly, P.D.; Gangell, C.L.; Chen, L.; Ware, R.S.; Ranganathan, S.; Mott, L.S.; Murray, C.P.; Stick, S.M. Risk factors for bronchiectasis in children with cystic fibrosis. N. Engl. J. Med. 2013 , 368 , 1963–1970. [ Google Scholar ] [ CrossRef ]
- Stoltz, D.A.; Meyerholz, D.K.; Pezzulo, A.A.; Ramachandran, S.; Rogan, M.P.; Davis, G.J.; Hanfland, R.A.; Wohlford-Lenane, C.; Dohrn, C.L.; Bartlett, J.A.; et al. Cystic fibrosis pigs develop lung disease and exhibit defective bacterial eradication at birth. Sci. Transl. Med. 2010 , 2 , 29ra31. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Stoltz, D.A.; Meyerholz, D.K.; Welsh, M.J. Origins of cystic fibrosis lung disease. N. Engl. J. Med. 2015 , 372 , 351–362. [ Google Scholar ] [ CrossRef ]
- Sun, X.; Olivier, A.K.; Liang, B.; Yi, Y.; Sui, H.; Evans, T.I.; Zhang, Y.; Zhou, W.; Tyler, S.R.; Fisher, J.T.; et al. Lung phenotype of juvenile and adult cystic fibrosis transmembrane conductance regulator-knockout ferrets. Am. J. Respir. Cell Mol. Biol. 2014 , 50 , 502–512. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Kelly, T.; Buxbaum, J. Gastrointestinal manifestations of cystic fibrosis. Dig. Dis. Sci. 2015 , 60 , 1903–1913. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Kopelman, H.; Durie, P.; Gaskin, K.; Weizman, Z.; Forstner, G. Pancreatic fluid secretion and protein hyperconcentration in cystic fibrosis. N. Engl. J. Med. 1985 , 312 , 329–334. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Shwachman, H.; Lebenthal, E.; Khaw, K.-T. Recurrent acute pancreatitis in patients with cystic fibrosis with normal pancreatic enzymes. Pediatrics 1975 , 55 , 86–95. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Waters, D.L.; Dorney, S.F.; Gaskin, K.J.; Gruca, M.A.; O’Halloran, M.; Wilcken, B. Pancreatic function in infants identified as having cystic fibrosis in a neonatal screening program. N. Engl. J. Med. 1990 , 322 , 303–308. [ Google Scholar ] [ CrossRef ]
- McKay, I.R.; Ooi, C.Y. The exocrine pancreas in cystic fibrosis in the era of CFTR modulation: A mini review. Front. Pediatr. 2022 , 10 , 914790. [ Google Scholar ] [ CrossRef ]
- Flume, P.A.; Robinson, K.A.; O’Sullivan, B.P.; Finder, J.D.; Vender, R.L.; Willey-Courand, D.-B.; White, T.B.; Marshall, B.C.; Committee, C.P.G.f.P.T. Cystic fibrosis pulmonary guidelines: Airway clearance therapies. Respir. Care 2009 , 54 , 522–537. [ Google Scholar ]
- Ritivoiu, M.-E.; Drăgoi, C.M.; Matei, D.; Stan, I.V.; Nicolae, A.C.; Craiu, M.; Dumitrescu, I.-B.; Ciolpan, A.A. Current and Future Therapeutic Approaches of Exocrine Pancreatic Insufficiency in Children with Cystic Fibrosis in the Era of Personalized Medicine. Pharmaceutics 2023 , 15 , 162. [ Google Scholar ] [ CrossRef ]
- Gibson-Corley, K.N.; Meyerholz, D.K.; Engelhardt, J.F. Pancreatic pathophysiology in cystic fibrosis. J. Pathol. 2016 , 238 , 311–320. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Kayani, K.; Mohammed, R.; Mohiaddin, H. Cystic fibrosis-related diabetes. Front. Endocrinol. 2018 , 9 , 331856. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Sabharwal, S. Gastrointestinal manifestations of cystic fibrosis. Gastroenterol. Hepatol. 2016 , 12 , 43. [ Google Scholar ]
- Van der Doef, H.P.; Kokke, F.; van der Ent, C.K.; Houwen, R.H. Intestinal obstruction syndromes in cystic fibrosis: Meconium ileus, distal intestinal obstruction syndrome, and constipation. Curr. Gastroenterol. Rep. 2011 , 13 , 265–270. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Kerem, E.; Corey, M.; Kerem, B.; Durie, P.; Tsui, L.C.; Levison, H. Clinical and genetic comparisons of patients with cystic fibrosis, with or without meconium ileus. J. Pediatr. 1989 , 114 , 767–773. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Chaudry, G.; Navarro, O.M.; Levine, D.S.; Oudjhane, K. Abdominal manifestations of cystic fibrosis in children. Pediatr. Radiol. 2006 , 36 , 233–240. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Lang, I.; Daneman, A.; Cutz, E.; Hagen, P.; Shandling, B. Abdominal calcification in cystic fibrosis with meconium ileus: Radiologic-pathologic correlation. Pediatr. Radiol. 1997 , 27 , 523–527. [ Google Scholar ] [ CrossRef ]
- Del Pin, C.A.; Czyrko, C.; Ziegler, M.M.; Scanlin, T.F.; Bishop, H.C. Management and survival of meconium ileus. A 30-year review. Ann. Surg. 1992 , 215 , 179. [ Google Scholar ] [ CrossRef ]
- Constantine, S.; Au, V.; Slavotinek, J. Abdominal manifestations of cystic fibrosis in adults: A review. Australas. Radiol. 2004 , 48 , 450–458. [ Google Scholar ] [ CrossRef ]
- McCarthy, V.A.; Harris, A. The CFTR gene and regulation of its expression. Pediatr. Pulmonol. 2005 , 40 , 1–8. [ Google Scholar ] [ CrossRef ]
- Zielenski, J.; Rozmahel, R.; Bozon, D.; Kerem, B.-s.; Grzelczak, Z.; Riordan, J.R.; Rommens, J.; Tsui, L.-C. Genomic DNA sequence of the cystic fibrosis transmembrane conductance regulator (CFTR) gene. Genomics 1991 , 10 , 214–228. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Rommens, J.M.; Iannuzzi, M.C.; Kerem, B.-s.; Drumm, M.L.; Melmer, G.; Dean, M.; Rozmahel, R.; Cole, J.L.; Kennedy, D.; Hidaka, N. Identification of the cystic fibrosis gene: Chromosome walking and jumping. Science 1989 , 245 , 1059–1065. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- CFTR2. Cystic Fibrosis Foundation, John Hopkins Medicine ; Sequenom Laboratories: San Diego, CA, SUA, 2023. [ Google Scholar ]
- Bobadilla, J.L.; Macek, M., Jr.; Fine, J.P.; Farrell, P.M. Cystic fibrosis: A worldwide analysis of CFTR mutations—Correlation with incidence data and application to screening. Hum. Mutat. 2002 , 19 , 575–606. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Marson, F.A.L.; Bertuzzo, C.S.; Ribeiro, J.D. Classification of CFTR mutation classes. Lancet Respir. Med. 2016 , 4 , e37–e38. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Parisi, G.F.; Mòllica, F.; Giallongo, A.; Papale, M.; Manti, S.; Leonardi, S. Cystic fibrosis transmembrane conductance regulator (CFTR): Beyond cystic fibrosis. Egypt. J. Med. Hum. Genet. 2022 , 23 , 94. [ Google Scholar ] [ CrossRef ]
- Elborn, J.S. Cystic fibrosis. Lancet 2016 , 388 , 2519–2531. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Veit, G.; Avramescu, R.G.; Chiang, A.N.; Houck, S.A.; Cai, Z.; Peters, K.W.; Hong, J.S.; Pollard, H.B.; Guggino, W.B.; Balch, W.E. From CFTR biology toward combinatorial pharmacotherapy: Expanded classification of cystic fibrosis mutations. Mol. Biol. Cell 2016 , 27 , 424–433. [ Google Scholar ] [ CrossRef ]
- Amaral, M.D. CFTR and chaperones. J. Mol. Neurosci. 2004 , 23 , 41–48. [ Google Scholar ] [ CrossRef ]
- Tsui, L.-C. The cystic fibrosis transmembrane conductance regulator gene. Am. J. Respir. Crit. Care Med. 1995 , 151 , S47. [ Google Scholar ] [ CrossRef ]
- Roxo-Rosa, M.; Xu, Z.; Schmidt, A.; Neto, M.; Cai, Z.; Soares, C.M.; Sheppard, D.N.; Amaral, M.D. Revertant mutants G550E and 4RK rescue cystic fibrosis mutants in the first nucleotide-binding domain of CFTR by different mechanisms. Proc. Natl. Acad. Sci. USA 2006 , 103 , 17891–17896. [ Google Scholar ] [ CrossRef ]
- Gregory, R.J.; Rich, D.P.; Cheng, S.H.; Souza, D.; Paul, S.; Manavalan, P.; Anderson, M.P.; Welsh, M.J.; Smith, A.E. Maturation and function of cystic fibrosis transmembrane conductance regulator variants bearing mutations in putative nucleotide-binding domains 1 and 2. Mol. Cell. Biol. 1991 , 11 , 3886–3893. [ Google Scholar ] [ PubMed ]
- Seibert, F.S.; Linsdell, P.; Loo, T.W.; Hanrahan, J.W.; Clarke, D.M.; Riordan, J.R. Disease-associated mutations in the fourth cytoplasmic loop of cystic fibrosis transmembrane conductance regulator compromise biosynthetic processing and chloride channel activity. J. Biol. Chem. 1996 , 271 , 15139–15145. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Bell, S.C.; De Boeck, K.; Amaral, M.D. New pharmacological approaches for cystic fibrosis: Promises, progress, pitfalls. Pharmacol. Ther. 2014 , 145 , 19–34. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Bradbury, N.A. CFTR and cystic fibrosis: A need for personalized medicine. In Studies of Epithelial Transporters and Ion Channels ; Springer: New York, NY, USA, 2020; pp. 547–604. [ Google Scholar ]
- Lopes-Pacheco, M. CFTR modulators: Shedding light on precision medicine for cystic fibrosis. Front. Pharmacol. 2016 , 7 , 275. [ Google Scholar ] [ CrossRef ]
- Ramalho, A.S.; Beck, S.; Meyer, M.; Penque, D.; Cutting, G.R.; Amaral, M.D. Five percent of normal cystic fibrosis transmembrane conductance regulator mRNA ameliorates the severity of pulmonary disease in cystic fibrosis. Am. J. Respir. Cell Mol. Biol. 2002 , 27 , 619–627. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Silvis, M.R.; Picciano, J.A.; Bertrand, C.; Weixel, K.; Bridges, R.J.; Bradbury, N.A. A mutation in the cystic fibrosis transmembrane conductance regulator generates a novel internalization sequence and enhances endocytic rates. J. Biol. Chem. 2003 , 278 , 11554–11560. [ Google Scholar ] [ CrossRef ]
- Haardt, M.; Benharouga, M.; Lechardeur, D.; Kartner, N.; Lukacs, G.L. C-terminal truncations destabilize the cystic fibrosis transmembrane conductance regulator without impairing its biogenesis: A novel class of mutation. J. Biol. Chem. 1999 , 274 , 21873–21877. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Lukacs, G.; Chang, X.-B.; Bear, C.; Kartner, N.; Mohamed, A.; Riordan, J.; Grinstein, S. The delta F508 mutation decreases the stability of cystic fibrosis transmembrane conductance regulator in the plasma membrane. Determination of functional half-lives on transfected cells. J. Biol. Chem. 1993 , 268 , 21592–21598. [ Google Scholar ] [ CrossRef ]
- Gentzsch, M.; Riordan, J.R. Localization of sequences within the C-terminal domain of the cystic fibrosis transmembrane conductance regulator which impact maturation and stability. J. Biol. Chem. 2001 , 276 , 1291–1298. [ Google Scholar ] [ CrossRef ]
- Benharouga, M.; Haardt, M.; Kartner, N.; Lukacs, G.L. COOH-terminal truncations promote proteasome-dependent degradation of mature cystic fibrosis transmembrane conductance regulator from post-Golgi compartments. J. Cell Biol. 2001 , 153 , 957–970. [ Google Scholar ] [ CrossRef ]
- Ramalho, A.S.; Lewandowska, M.A.; Farinha, C.M.; Mendes, F.; Gonçalves, J.; Barreto, C.; Harris, A.; Amaral, M.D. Deletion of CFTR translation start site reveals functional isoforms of the protein in CF patients. Cell. Physiol. Biochem. 2009 , 24 , 335–346. [ Google Scholar ] [ CrossRef ]
- Farinha, C.M.; King-Underwood, J.; Sousa, M.; Correia, A.R.; Henriques, B.J.; Roxo-Rosa, M.; Da Paula, A.C.; Williams, J.; Hirst, S.; Gomes, C.M. Revertants, low temperature, and correctors reveal the mechanism of F508del-CFTR rescue by VX-809 and suggest multiple agents for full correction. Chem. Biol. 2013 , 20 , 943–955. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- He, L.; Kota, P.; Aleksandrov, A.A.; Cui, L.; Jensen, T.; Dokholyan, N.V.; Riordan, J.R. Correctors of ΔF508 CFTR restore global conformational maturation without thermally stabilizing the mutant protein. FASEB J. 2013 , 27 , 536–545. [ Google Scholar ] [ CrossRef ]
- Pereira, S.V.-N.; Ribeiro, J.D.; Ribeiro, A.F.; Bertuzzo, C.S.; Marson, F.A.L. Novel, rare and common pathogenic variants in the CFTR gene screened by high-throughput sequencing technology and predicted by in silico tools. Sci. Rep. 2019 , 9 , 6234. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Deletang, K.; Taulan-Cadars, M. Splicing mutations in the CFTR gene as therapeutic targets. Gene Ther. 2022 , 29 , 399–406. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Nagel, G.; Hwang, T.-C.; Nastiuk, K.L.; Nairn, A.C.; Gadsbyt, D.C. The protein kinase A-regulated cardiac CI − channel resembles the cystic fibrosis transmembrane conductance regulator. Nature 1992 , 360 , 81–84. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Chang, X.B.; Hou, Y.X.; Jensen, T.J.; Riordan, J.R. Mapping of cystic fibrosis transmembrane conductance regulator membrane topology by glycosylation site insertion. J. Biol. Chem. 1994 , 269 , 18572–18575. [ Google Scholar ] [ CrossRef ]
- Li, C.; Naren, A.P. Macromolecular complexes of cystic fibrosis transmembrane conductance regulator and its interacting partners. Pharmacol. Ther. 2005 , 108 , 208–223. [ Google Scholar ] [ CrossRef ]
- Aleksandrov, A.A.; Kota, P.; Aleksandrov, L.A.; He, L.; Jensen, T.; Cui, L.; Gentzsch, M.; Dokholyan, N.V.; Riordan, J.R. Regulatory insertion removal restores maturation, stability and function of DeltaF508 CFTR. J. Mol. Biol. 2010 , 401 , 194–210. [ Google Scholar ] [ CrossRef ]
- Cheng, S.H.; Rich, D.P.; Marshall, J.; Gregory, R.J.; Welsh, M.J.; Smith, A.E. Phosphorylation of the R domain by cAMP-dependent protein kinase regulates the CFTR chloride channel. Cell 1991 , 66 , 1027–1036. [ Google Scholar ] [ CrossRef ]
- Anderson, M.P.; Berger, H.A.; Rich, D.P.; Gregory, R.J.; Smith, A.E.; Welsh, M.J. Nucleoside triphosphates are required to open the CFTR chloride channel. Cell 1991 , 67 , 775–784. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Csanády, L.; Vergani, P.; Gadsby, D.C. Structure, gating, and regulation of the CFTR anion channel. Physiol. Rev. 2019 , 99 , 707–738. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Hallows, K.R.; Raghuram, V.; Kemp, B.E.; Witters, L.A.; Foskett, J.K. Inhibition of cystic fibrosis transmembrane conductance regulator by novel interaction with the metabolic sensor AMP-activated protein kinase. J. Clin. Investig. 2000 , 105 , 1711–1721. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Kunzelmann, K.; Mehta, A. CFTR: A hub for kinases and crosstalk of c AMP and Ca 2+ . FEBS J. 2013 , 280 , 4417–4429. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Luo, J.; Pato, M.D.; Riordan, J.R.; Hanrahan, J.W. Differential regulation of single CFTR channels by PP2C, PP2A, and other phosphatases. Am. J. Physiol. Cell Physiol. 1998 , 274 , C1397–C1410. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Vergani, P.; Lockless, S.W.; Nairn, A.C.; Gadsby, D.C. CFTR channel opening by ATP-driven tight dimerization of its nucleotide-binding domains. Nature 2005 , 433 , 876–880. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Naren, A.P. Methods for the study of intermolecular and intramolecular interactions regulating CFTR function. In Cystic Fibrosis Methods and Protocols ; Springer: New York, NY, USA, 2002; pp. 175–186. [ Google Scholar ]
- Wilkinson, D.J.; Strong, T.V.; Mansoura, M.K.; Wood, D.L.; Smith, S.S.; Collins, F.S.; Dawson, D.C. CFTR activation: Additive effects of stimulatory and inhibitory phosphorylation sites in the R domain. Am. J. Physiol. Lung Cell. Mol. Physiol. 1997 , 273 , L127–L133. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Schreiber, R.; Nitschke, R.; Greger, R.; Kunzelmann, K. The cystic fibrosis transmembrane conductance regulator activates aquaporin 3 in airway epithelial cells. J. Biol. Chem. 1999 , 274 , 11811–11816. [ Google Scholar ] [ CrossRef ]
- Boucher, R.C.; Stutts, M.J.; Knowles, M.R.; Cantley, L.; Gatzy, J.T. Na + transport in cystic fibrosis respiratory epithelia. Abnormal basal rate and response to adenylate cyclase activation. J. Clin. Investig. 1986 , 78 , 1245–1252. [ Google Scholar ] [ CrossRef ]
- McNicholas, C.M.; Guggino, W.B.; Schwiebert, E.M.; Hebert, S.C.; Giebisch, G.; Egan, M.E. Sensitivity of a renal K + channel (ROMK2) to the inhibitory sulfonylurea compound glibenclamide is enhanced by coexpression with the ATP-binding cassette transporter cystic fibrosis transmembrane regulator. Proc. Natl. Acad. Sci. USA 1996 , 93 , 8083–8088. [ Google Scholar ] [ CrossRef ]
- Gabriel, S.E.; Clarke, L.L.; Boucher, R.C.; Stutts, M.J. CFTR and outward rectifying chloride channels are distinct proteins with a regulatory relationship. Nature 1993 , 363 , 263–266. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Lee, M.G.; Wigley, W.C.; Zeng, W.; Noel, L.E.; Marino, C.R.; Thomas, P.J.; Muallem, S. Regulation of Cl − /HCO 3− exchange by cystic fibrosis transmembrane conductance regulator expressed in NIH 3T3 and HEK 293 cells. J. Biol. Chem. 1999 , 274 , 3414–3421. [ Google Scholar ] [ CrossRef ]
- Zhang, Z.; Liu, F.; Chen, J. Molecular structure of the ATP-bound, phosphorylated human CFTR. Proc. Natl. Acad. Sci. USA 2018 , 115 , 12757–12762. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Hwang, T.-C.; Yeh, J.-T.; Zhang, J.; Yu, Y.-C.; Yeh, H.-I.; Destefano, S. Structural mechanisms of CFTR function and dysfunction. J. Gen. Physiol. 2018 , 150 , 539–570. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Rosenberg, M.F.; Kamis, A.B.; Aleksandrov, L.A.; Ford, R.C.; Riordan, J.R. Purification and crystallization of the cystic fibrosis transmembrane conductance regulator (CFTR). J. Biol. Chem. 2004 , 279 , 39051–39057. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Zhang, Z.; Chen, J. Atomic Structure of the Cystic Fibrosis Transmembrane Conductance Regulator. Cell 2016 , 167 , 1586–1597.e1589. [ Google Scholar ] [ CrossRef ]
- Mihályi, C.; Töröcsik, B.; Csanády, L. Obligate coupling of CFTR pore opening to tight nucleotide-binding domain dimerization. Elife 2016 , 5 , e18164. [ Google Scholar ] [ CrossRef ]
- Mihályi, C.; Iordanov, I.; Töröcsik, B.; Csanády, L. Simple binding of protein kinase A prior to phosphorylation allows CFTR anion channels to be opened by nucleotides. Proc. Natl. Acad. Sci. USA 2020 , 117 , 21740–21746. [ Google Scholar ] [ CrossRef ]
- Naren, A.P.; Nelson, D.J.; Xie, W.; Jovov, B.; Pevsner, J.; Bennett, M.K.; Benos, D.J.; Quick, M.W.; Kirk, K.L. Regulation of CFTR chloride channels by syntaxin and Munc18 isoforms. Nature 1997 , 390 , 302–305. [ Google Scholar ] [ CrossRef ]
- Naren, A.P.; Cormet-Boyaka, E.; Fu, J.; Villain, M.; Blalock, J.E.; Quick, M.W.; Kirk, K.L. CFTR chloride channel regulation by an interdomain interaction. Science 1999 , 286 , 544–548. [ Google Scholar ] [ CrossRef ]
- Scotet, V.; L’Hostis, C.; Férec, C. The Changing Epidemiology of Cystic Fibrosis: Incidence, Survival and Impact of the CFTR Gene Discovery. Genes 2020 , 11 , 589. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Van Goor, F.; Hadida, S.; Grootenhuis, P.D.; Burton, B.; Cao, D.; Neuberger, T.; Turnbull, A.; Singh, A.; Joubran, J.; Hazlewood, A. Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc. Natl. Acad. Sci. USA 2009 , 106 , 18825–18830. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Goor, F.V.; Straley, K.S.; Cao, D.; González, J.; Hadida, S.; Hazlewood, A.; Joubran, J.; Knapp, T.; Makings, L.R.; Miller, M.; et al. Rescue of ΔF508-CFTR trafficking and gating in human cystic fibrosis airway primary cultures by small molecules. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006 , 290 , L1117–L1130. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Fukuda, R.; Okiyoneda, T. Peripheral protein quality control as a novel drug target for CFTR stabilizer. Front. Pharmacol. 2018 , 9 , 1100. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Giuliano, K.A.; Wachi, S.; Drew, L.; Dukovski, D.; Green, O.; Bastos, C.; Cullen, M.D.; Hauck, S.; Tait, B.D.; Munoz, B. Use of a high-throughput phenotypic screening strategy to identify amplifiers, a novel pharmacological class of small molecules that exhibit functional synergy with potentiators and correctors. SLAS Discovery. 2018 , 23 , 111–121. [ Google Scholar ] [ CrossRef ]
- Clancy, J.P.; Cotton, C.U.; Donaldson, S.H.; Solomon, G.M.; VanDevanter, D.R.; Boyle, M.P.; Gentzsch, M.; Nick, J.A.; Illek, B.; Wallenburg, J.C.; et al. CFTR modulator theratyping: Current status, gaps and future directions. J. Cyst. Fibros. 2019 , 18 , 22–34. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Bear, C.E. A therapy for most with cystic fibrosis. Cell 2020 , 180 , 211. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Davis, P.B.; Yasothan, U.; Kirkpatrick, P. Ivacaftor. Nat. Rev. Drug Discov. 2012 , 11 , 349–351. [ Google Scholar ] [ CrossRef ]
- Wainwright, C.E.; Elborn, J.S.; Ramsey, B.W.; Marigowda, G.; Huang, X.; Cipolli, M.; Colombo, C.; Davies, J.C.; De Boeck, K.; Flume, P.A. Lumacaftor–ivacaftor in patients with cystic fibrosis homozygous for Phe508del CFTR. N. Engl. J. Med. 2015 , 373 , 220–231. [ Google Scholar ] [ CrossRef ]
- Cholon, D.M.; Quinney, N.L.; Fulcher, M.L.; Esther, C.R., Jr.; Das, J.; Dokholyan, N.V.; Randell, S.H.; Boucher, R.C.; Gentzsch, M. Potentiator ivacaftor abrogates pharmacological correction of ΔF508 CFTR in cystic fibrosis. Sci. Transl. Med. 2014 , 6 , 246ra96. [ Google Scholar ] [ CrossRef ]
- Sala, M.A.; Jain, M. Tezacaftor for the treatment of cystic fibrosis. Expert Rev. Respir. Med. 2018 , 12 , 725–732. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Middleton, P.G.; Mall, M.A.; Dřevínek, P.; Lands, L.C.; McKone, E.F.; Polineni, D.; Ramsey, B.W.; Taylor-Cousar, J.L.; Tullis, E.; Vermeulen, F. Elexacaftor–tezacaftor–ivacaftor for cystic fibrosis with a single Phe508del allele. N. Engl. J. Med. 2019 , 381 , 1809–1819. [ Google Scholar ] [ CrossRef ]
- Ramsey, B.W.; Davies, J.; McElvaney, N.G.; Tullis, E.; Bell, S.C.; Dřevínek, P.; Griese, M.; McKone, E.F.; Wainwright, C.E.; Konstan, M.W. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N. Engl. J. Med. 2011 , 365 , 1663–1672. [ Google Scholar ] [ CrossRef ]
- Becq, F.; Mirval, S.; Carrez, T.; Lévêque, M.; Billet, A.; Coraux, C.; Sage, E.; Cantereau, A. The rescue of F508del-CFTR by elexacaftor/tezacaftor/ivacaftor (Trikafta) in human airway epithelial cells is underestimated due to the presence of ivacaftor. Eur. Respir. J. 2022 , 59 , 2100671. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Keating, D.; Marigowda, G.; Burr, L.; Daines, C.; Mall, M.A.; McKone, E.F.; Ramsey, B.W.; Rowe, S.M.; Sass, L.A.; Tullis, E. VX-445–tezacaftor–ivacaftor in patients with cystic fibrosis and one or two Phe508del alleles. N. Engl. J. Med. 2018 , 379 , 1612–1620. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Heijerman, H.G.; McKone, E.F.; Downey, D.G.; Van Braeckel, E.; Rowe, S.M.; Tullis, E.; Mall, M.A.; Welter, J.J.; Ramsey, B.W.; McKee, C.M. Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: A double-blind, randomised, phase 3 trial. Lancet 2019 , 394 , 1940–1948. [ Google Scholar ] [ CrossRef ]
- Bell, S.C.; Barry, P.J.; De Boeck, K.; Drevinek, P.; Elborn, J.S.; Plant, B.J.; Minić, P.; Van Braeckel, E.; Verhulst, S.; Muller, K. CFTR activity is enhanced by the novel corrector GLPG2222, given with and without ivacaftor in two randomized trials. J. Cyst. Fibros. 2019 , 18 , 700–707. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Liu, F.; Zhang, Z.; Levit, A.; Levring, J.; Touhara, K.K.; Shoichet, B.K.; Chen, J. Structural identification of a hotspot on CFTR for potentiation. Science 2019 , 364 , 1184–1188. [ Google Scholar ] [ CrossRef ]
- Sinha, C.; Zhang, W.; Moon, C.S.; Actis, M.; Yarlagadda, S.; Arora, K.; Woodroofe, K.; Clancy, J.P.; Lin, S.; Ziady, A.G.; et al. Capturing the Direct Binding of CFTR Correctors to CFTR by Using Click Chemistry. Chembiochem 2015 , 16 , 2017–2022. [ Google Scholar ] [ CrossRef ]
- Fiedorczuk, K.; Chen, J. Mechanism of CFTR correction by type I folding correctors. Cell 2022 , 185 , 158–168.e11. [ Google Scholar ] [ CrossRef ]
- Fiedorczuk, K.; Chen, J. Molecular structures reveal synergistic rescue of Δ508 CFTR by Trikafta modulators. Science 2022 , 378 , 284–290. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Eckford, P.D.; Li, C.; Ramjeesingh, M.; Bear, C.E. Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Potentiator VX-770 (Ivacaftor) Opens the Defective Channel Gate of Mutant CFTR in a Phosphorylation-dependent but ATP-independent Manner. J. Biol. Chem. 2012 , 287 , 36639–36649. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Cui, G.; Stauffer, B.B.; Imhoff, B.R.; Rab, A.; Hong, J.S.; Sorscher, E.J.; McCarty, N.A. VX-770-mediated potentiation of numerous human CFTR disease mutants is influenced by phosphorylation level. Sci. Rep. 2019 , 9 , 13460. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Loo, T.W.; Bartlett, M.C.; Clarke, D.M. Corrector VX-809 stabilizes the first transmembrane domain of CFTR. Biochem. Pharmacol. 2013 , 86 , 612–619. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Okiyoneda, T.; Veit, G.; Dekkers, J.F.; Bagdany, M.; Soya, N.; Xu, H.; Roldan, A.; Verkman, A.S.; Kurth, M.; Simon, A. Mechanism-based corrector combination restores ΔF508-CFTR folding and function. Nat. Chem. Biol. 2013 , 9 , 444–454. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Laselva, O.; Molinski, S.; Casavola, V.; Bear, C.E. Correctors of the major cystic fibrosis mutant interact through membrane-spanning domains. Mol. Pharmacol. 2018 , 93 , 612–618. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Eckford, P.D.; Ramjeesingh, M.; Molinski, S.; Pasyk, S.; Dekkers, J.F.; Li, C.; Ahmadi, S.; Ip, W.; Chung, T.E.; Du, K. VX-809 and related corrector compounds exhibit secondary activity stabilizing active F508del-CFTR after its partial rescue to the cell surface. Chem. Biol. 2014 , 21 , 666–678. [ Google Scholar ] [ CrossRef ]
- Pedemonte, N.; Bertozzi, F.; Caci, E.; Sorana, F.; Di Fruscia, P.; Tomati, V.; Ferrera, L.; Rodríguez-Gimeno, A.; Berti, F.; Pesce, E. Discovery of a picomolar potency pharmacological corrector of the mutant CFTR chloride channel. Sci. Adv. 2020 , 6 , eaay9669. [ Google Scholar ] [ CrossRef ]
- Wang, X.; Liu, B.; Searle, X.; Yeung, C.; Bogdan, A.; Greszler, S.; Singh, A.; Fan, Y.; Swensen, A.M.; Vortherms, T. Discovery of 4-[(2R,4R)-4-({[1-(2,2-Difluoro-1,3-benzodioxol-5-yl)cyclopropyl]carbonyl}amino)-7-(difluoromethoxy)-3,4-dihydro-2H-chromen-2-yl]benzoic Acid(ABBV/GLPG-2222), a Potent Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Corrector for the Treatment of Cystic Fibrosis ; ACS Publications: Washington, DC, USA, 2018. [ Google Scholar ]
- Veit, G.; Xu, H.; Dreano, E.; Avramescu, R.G.; Bagdany, M.; Beitel, L.K.; Roldan, A.; Hancock, M.A.; Lay, C.; Li, W.; et al. Structure-guided combination therapy to potently improve the function of mutant CFTRs. Nat. Med. 2018 , 24 , 1732–1742. [ Google Scholar ] [ CrossRef ]
- Scanio, M.J.; Searle, X.B.; Liu, B.; Koenig, J.R.; Altenbach, R.; Gfesser, G.A.; Bogdan, A.; Greszler, S.; Zhao, G.; Singh, A. Discovery of ABBV/GLPG-3221, a Potent Corrector of CFTR for the Treatment of Cystic Fibrosis. ACS Med. Chem. Lett. 2019 , 10 , 1543–1548. [ Google Scholar ] [ CrossRef ]
- De Wilde, G.; Gees, M.; Musch, S.; Verdonck, K.; Jans, M.; Wesse, A.-S.; Singh, A.K.; Hwang, T.-C.; Christophe, T.; Pizzonero, M. Identification of GLPG/ABBV-2737, a novel class of corrector, which exerts functional synergy with other CFTR modulators. Front. Pharmacol. 2019 , 10 , 514. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Veit, G.; Roldan, A.; Hancock, M.A.; Da Fonte, D.F.; Xu, H.; Hussein, M.; Frenkiel, S.; Matouk, E.; Velkov, T.; Lukacs, G.L. Allosteric folding correction of F508del and rare CFTR mutants by elexacaftor-tezacaftor-ivacaftor (Trikafta) combination. JCI Insight 2020 , 5 , e139983. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Burgener, E.B.; Cornfield, D.N. Delivering a New Future for People with Cystic Fibrosis. Pediatrics 2023 , 152 , e2023062985. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Ong, T.; Ramsey, B.W. New Therapeutic Approaches to Modulate and Correct Cystic Fibrosis Transmembrane Conductance Regulator. Pediatr. Clin. N. Am. 2016 , 63 , 751–764. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Mort, M.; Ivanov, D.; Cooper, D.N.; Chuzhanova, N.A. A meta-analysis of nonsense mutations causing human genetic disease. Hum. Mutat. 2008 , 29 , 1037–1047. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- De Boeck, K.; Zolin, A.; Cuppens, H.; Olesen, H.V.; Viviani, L. The relative frequency of CFTR mutation classes in European patients with cystic fibrosis. J. Cyst. Fibros. 2014 , 13 , 403–409. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Keeling, K.M.; Xue, X.; Gunn, G.; Bedwell, D.M. Therapeutics based on stop codon readthrough. Annu. Rev. Genom. Hum. Genet. 2014 , 15 , 371–394. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Forge, A.; Schacht, J. Aminoglycoside Antibiotics. Audiol. Neurotol. 2000 , 5 , 3–22. [ Google Scholar ] [ CrossRef ]
- Sharma, J.; Du, M.; Wong, E.; Mutyam, V.; Li, Y.; Chen, J.; Wangen, J.; Thrasher, K.; Fu, L.; Peng, N.; et al. A small molecule that induces translational readthrough of CFTR nonsense mutations by eRF1 depletion. Nat. Commun. 2021 , 12 , 4358. [ Google Scholar ] [ CrossRef ]
- Konstan, M.W.; VanDevanter, D.R.; Rowe, S.M.; Wilschanski, M.; Kerem, E.; Sermet-Gaudelus, I.; DiMango, E.; Melotti, P.; McIntosh, J.; De Boeck, K. Efficacy and safety of ataluren in patients with nonsense-mutation cystic fibrosis not receiving chronic inhaled aminoglycosides: The international, randomized, double-blind, placebo-controlled Ataluren Confirmatory Trial in Cystic Fibrosis (ACT CF). J. Cyst. Fibros. 2020 , 19 , 595–601. [ Google Scholar ] [ CrossRef ]
- Crawford, D.K.; Mullenders, J.; Pott, J.; Boj, S.F.; Landskroner-Eiger, S.; Goddeeris, M.M. Targeting G542X CFTR nonsense alleles with ELX-02 restores CFTR function in human-derived intestinal organoids. J. Cyst. Fibros. 2021 , 20 , 436–442. [ Google Scholar ] [ CrossRef ]
- Aksit, M.A.; Bowling, A.D.; Evans, T.A.; Joynt, A.T.; Osorio, D.; Patel, S.; West, N.; Merlo, C.; Sosnay, P.R.; Cutting, G.R.; et al. Decreased mRNA and protein stability of W1282X limits response to modulator therapy. J. Cyst. Fibros. 2019 , 18 , 606–613. [ Google Scholar ] [ CrossRef ]
- Lee, R.E.; Lewis, C.A.; He, L.; Bulik-Sullivan, E.C.; Gallant, S.C.; Mascenik, T.M.; Dang, H.; Cholon, D.M.; Gentzsch, M.; Morton, L.C.; et al. Small-molecule eRF3a degraders rescue CFTR nonsense mutations by promoting premature termination codon readthrough. J. Clin. Investig. 2022 , 132 , e154571. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Baradaran-Heravi, A.; Balgi, A.D.; Hosseini-Farahabadi, S.; Choi, K.; Has, C.; Roberge, M. Effect of small molecule eRF3 degraders on premature termination codon readthrough. Nucleic Acids Res. 2021 , 49 , 3692–3708. [ Google Scholar ] [ CrossRef ]
- Pedemonte, N.; Sonawane, N.D.; Taddei, A.; Hu, J.; Zegarra-Moran, O.; Suen, Y.F.; Robins, L.I.; Dicus, C.W.; Willenbring, D.; Nantz, M.H.; et al. Phenylglycine and sulfonamide correctors of defective delta F508 and G551D cystic fibrosis transmembrane conductance regulator chloride-channel gating. Mol. Pharmacol. 2005 , 67 , 1797–1807. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Renda, M.; Barreca, M.; Borrelli, A.; Spanò, V.; Montalbano, A.; Raimondi, M.V.; Bivacqua, R.; Musante, I.; Scudieri, P.; Guidone, D.; et al. Novel tricyclic pyrrolo-quinolines as pharmacological correctors of the mutant CFTR chloride channel. Sci. Rep. 2023 , 13 , 7604. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Laselva, O.; Eckford, P.D.; Bartlett, C.; Ouyang, H.; Gunawardena, T.N.; Gonska, T.; Moraes, T.J.; Bear, C.E. Functional rescue of c. 3846G> A (W1282X) in patient-derived nasal cultures achieved by inhibition of nonsense mediated decay and protein modulators with complementary mechanisms of action. J. Cyst. Fibros. 2020 , 19 , 717–727. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Laselva, O.; Bartlett, C.; Popa, A.; Ouyang, H.; Gunawardena, T.N.; Gonska, T.; Moraes, T.J.; Bear, C.E. Emerging preclinical modulators developed for F508del-CFTR have the potential to be effective for ORKAMBI resistant processing mutants. J. Cyst. Fibros. 2021 , 20 , 106–119. [ Google Scholar ] [ CrossRef ]
- Laselva, O.; Guerra, L.; Castellani, S.; Favia, M.; Di Gioia, S.; Conese, M. Small-molecule drugs for cystic fibrosis: Where are we now? Pulm. Pharmacol. Ther. 2022 , 72 , 102098. [ Google Scholar ] [ CrossRef ]
- Mergiotti, M.; Murabito, A.; Prono, G.; Ghigo, A. CFTR Modulator Therapy for Rare CFTR Mutants. J. Respir. 2022 , 2 , 59–76. [ Google Scholar ] [ CrossRef ]
- Pranke, I.; Golec, A.; Hinzpeter, A.; Edelman, A.; Sermet-Gaudelus, I. Emerging Therapeutic Approaches for Cystic Fibrosis. From Gene Editing to Personalized Medicine. Front. Pharmacol. 2019 , 10 , 121. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Pringle, I.; Hyde, S.; Gill, D. Non-viral vectors in cystic fibrosis gene therapy: Recent developments and future prospects. Expert Opin. Biol. Ther. 2009 , 9 , 991–1003. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Gregory, L.G.; Harbottle, R.P.; Lawrence, L.; Knapton, H.J.; Themis, M.; Coutelle, C. Enhancement of adenovirus-mediated gene transfer to the airways by DEAE dextran and sodium caprate in vivo. Mol. Ther. 2003 , 7 , 19–26. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Scaria, A.; St. George, J.A.; Jiang, C.; Kaplan, J.M.; Wadsworth, S.C.; Gregory, R.J. Adenovirus-mediated persistent cystic fibrosis transmembrane conductance regulator expression in mouse airway epithelium. J. Virol. 1998 , 72 , 7302–7309. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Bellon, G.; Michel-Calemard, L.; Thouvenot, D.; Jagneaux, V.; Poitevin, F.; Malcus, C.; Accart, N.; Layani, M.P.; Aymard, M.; Bernon, H. Aerosol administration of a recombinant adenovirus expressing CFTR to cystic fibrosis patients: A phase I clinical trial. Hum. Gene Ther. 1997 , 8 , 15–25. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Hart, S.L.; Harrison, P.T. Genetic therapies for cystic fibrosis lung disease. Curr. Opin. Pharmacol. 2017 , 34 , 119–124. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Yan, Z.; Feng, Z.; Sun, X.; Zhang, Y.; Zou, W.; Wang, Z.; Jensen-Cody, C.; Liang, B.; Park, S.-Y.; Qiu, J. Human bocavirus type-1 capsid facilitates the transduction of ferret airways by adeno-associated virus genomes. Hum. Gene Ther. 2017 , 28 , 612–625. [ Google Scholar ] [ CrossRef ]
- Vidović, D.; Carlon, M.S.; Da Cunha, M.F.; Dekkers, J.F.; Hollenhorst, M.I.; Bijvelds, M.J.; Ramalho, A.S.; Van den Haute, C.; Ferrante, M.; Baekelandt, V. rAAV-CFTRΔR rescues the cystic fibrosis phenotype in human intestinal organoids and cystic fibrosis mice. Am. J. Respir. Crit. Care Med. 2016 , 193 , 288–298. [ Google Scholar ] [ CrossRef ]
- Foldvari, M.; Chen, D.W.; Nafissi, N.; Calderon, D.; Narsineni, L.; Rafiee, A. Non-viral gene therapy: Gains and challenges of non-invasive administration methods. J. Control. Release 2016 , 240 , 165–190. [ Google Scholar ] [ CrossRef ]
- Catanese, D.; Fogg, J.; Schrock, D.; Gilbert, B.; Zechiedrich, L. Supercoiled Minivector DNA resists shear forces associated with gene therapy delivery. Gene Ther. 2012 , 19 , 94–100. [ Google Scholar ] [ CrossRef ]
- Alton, E.W.; Armstrong, D.K.; Ashby, D.; Bayfield, K.J.; Bilton, D.; Bloomfield, E.V.; Boyd, A.C.; Brand, J.; Buchan, R.; Calcedo, R. Repeated nebulisation of non-viral CFTR gene therapy in patients with cystic fibrosis: A randomised, double-blind, placebo-controlled, phase 2b trial. Lancet Respir. Med. 2015 , 3 , 684–691. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Bangel-Ruland, N.; Tomczak, K.; Fernández Fernández, E.; Leier, G.; Leciejewski, B.; Rudolph, C.; Rosenecker, J.; Weber, W.M. Cystic fibrosis transmembrane conductance regulator-mRNA delivery: A novel alternative for cystic fibrosis gene therapy. J. Gene Med. 2013 , 15 , 414–426. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Robinson, E.; MacDonald, K.D.; Slaughter, K.; McKinney, M.; Patel, S.; Sun, C.; Sahay, G. Lipid nanoparticle-delivered chemically modified mRNA restores chloride secretion in cystic fibrosis. Mol. Ther. 2018 , 26 , 2034–2046. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Bennett, C.F. Therapeutic antisense oligonucleotides are coming of age. Annu. Rev. Med. 2019 , 70 , 307–321. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Zamecnik, P.C.; Raychowdhury, M.K.; Tabatadze, D.R.; Cantiello, H.F. Reversal of cystic fibrosis phenotype in a cultured Δ508 cystic fibrosis transmembrane conductance regulator cell line by oligonucleotide insertion. Proc. Natl. Acad. Sci. USA 2004 , 101 , 8150–8155. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Kim, Y.J.; Krainer, A.R. Antisense oligonucleotide therapeutics for cystic fibrosis: Recent developments and perspectives. Mol. Cells 2023 , 46 , 10–20. [ Google Scholar ] [ CrossRef ]
- Chiba-Falek, O.; Kerem, E.; Shoshani, T.; Aviram, M.; Augarten, A.; Bentur, L.; Tal, A.; Tullis, E.; Rahat, A.; Kerem, B. The molecular basis of disease variability among cystic fibrosis patients carrying the 3849 + 10 kb C → T mutation. Genomics 1998 , 53 , 276–283. [ Google Scholar ] [ CrossRef ]
- Friedman, K.J.; Kole, J.; Cohn, J.A.; Knowles, M.R.; Silverman, L.M.; Kole, R. Correction of aberrant splicing of the cystic fibrosis transmembrane conductance regulator (CFTR) gene by antisense oligonucleotides. J. Biol. Chem. 1999 , 274 , 36193–36199. [ Google Scholar ] [ CrossRef ]
- Michaels, W.E.; Pena-Rasgado, C.; Kotaria, R.; Bridges, R.J.; Hastings, M.L. Open reading frame correction using splice-switching antisense oligonucleotides for the treatment of cystic fibrosis. Proc. Natl. Acad. Sci. USA 2022 , 119 , e2114886119. [ Google Scholar ] [ CrossRef ]
- Zaher, A.; ElSaygh, J.; Elsori, D.; ElSaygh, H.; Sanni, A.; Elsaygh, H.; Sanni, A. A review of Trikafta: Triple cystic fibrosis transmembrane conductance regulator (CFTR) modulator therapy. Cureus 2021 , 13 , e16144. [ Google Scholar ] [ CrossRef ]
- Oren, Y.S.; Sinai, M.I.-T.; Golec, A.; Barchad-Avitzur, O.; Mutyam, V.; Li, Y.; Hong, J.; Ozeri-Galai, E.; Hatton, A.; Leibson, C. Antisense oligonucleotide-based drug development for Cystic Fibrosis patients carrying the 3849+10 kb C-to-T splicing mutation. J. Cyst. Fibros. 2021 , 20 , 865–875. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Crosby, J.R.; Zhao, C.; Jiang, C.; Bai, D.; Katz, M.; Greenlee, S.; Kawabe, H.; McCaleb, M.; Rotin, D.; Guo, S. Inhaled ENaC antisense oligonucleotide ameliorates cystic fibrosis-like lung disease in mice. J. Cyst. Fibros. 2017 , 16 , 671–680. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Schwank, G.; Koo, B.-K.; Sasselli, V.; Dekkers, J.F.; Heo, I.; Demircan, T.; Sasaki, N.; Boymans, S.; Cuppen, E.; van der Ent, C.K. Functional repair of CFTR by CRISPR/Cas9 in intestinal stem cell organoids of cystic fibrosis patients. Cell Stem Cell 2013 , 13 , 653–658. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Crane, A.M.; Kramer, P.; Bui, J.H.; Chung, W.J.; Li, X.S.; Gonzalez-Garay, M.L.; Hawkins, F.; Liao, W.; Mora, D.; Choi, S. Targeted correction and restored function of the CFTR gene in cystic fibrosis induced pluripotent stem cells. Stem Cell Rep. 2015 , 4 , 569–577. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Firth, A.L.; Menon, T.; Parker, G.S.; Qualls, S.J.; Lewis, B.M.; Ke, E.; Dargitz, C.T.; Wright, R.; Khanna, A.; Gage, F.H. Functional gene correction for cystic fibrosis in lung epithelial cells generated from patient iPSCs. Cell Rep. 2015 , 12 , 1385–1390. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Sanz, D.J.; Hollywood, J.A.; Scallan, M.F.; Harrison, P.T. Cas9/gRNA targeted excision of cystic fibrosis-causing deep-intronic splicing mutations restores normal splicing of CFTR mRNA. PLoS ONE 2017 , 12 , e0184009. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Guo, L.; Karoubi, G.; Duchesneau, P.; Shutova, M.V.; Sung, H.-K.; Tonge, P.; Bear, C.; Rogers, I.; Nagy, A.; Waddell, T.K. Generation of induced progenitor-like cells from mature epithelial cells using interrupted reprogramming. Stem Cell Rep. 2017 , 9 , 1780–1795. [ Google Scholar ] [ CrossRef ]
- Takahashi, K.; Okita, K.; Nakagawa, M.; Yamanaka, S. Induction of pluripotent stem cells from fibroblast cultures. Nat. Protoc. 2007 , 2 , 3081–3089. [ Google Scholar ] [ CrossRef ]
- Pollard, B.S.; Pollard, H.B. Induced pluripotent stem cells for treating cystic fibrosis: State of the science. Pediatr. Pulmonol. 2018 , 53 , S12–S29. [ Google Scholar ] [ CrossRef ]
- Schmidt, B.Z.; Haaf, J.B.; Leal, T.; Noel, S. Cystic fibrosis transmembrane conductance regulator modulators in cystic fibrosis: Current perspectives. Clin. Pharmacol. Adv. Appl. 2016 , 8 , 127–140. [ Google Scholar ]
- Simsek, S.; Zhou, T.; Robinson, C.L.; Tsai, S.-Y.; Crespo, M.; Amin, S.; Lin, X.; Hon, J.; Evans, T.; Chen, S. Modeling cystic fibrosis using pluripotent stem cell-derived human pancreatic ductal epithelial cells. Stem Cells Transl. Med. 2016 , 5 , 572–579. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Rock, J.R.; Onaitis, M.W.; Rawlins, E.L.; Lu, Y.; Clark, C.P.; Xue, Y.; Randell, S.H.; Hogan, B.L. Basal cells as stem cells of the mouse trachea and human airway epithelium. Proc. Natl. Acad. Sci. USA 2009 , 106 , 12771–12775. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Reid, L.M.; Jones, R. Mucous membrane of respiratory epithelium. Environ. Health Perspect. 1980 , 35 , 113–120. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Rawlins, E.L.; Okubo, T.; Xue, Y.; Brass, D.M.; Auten, R.L.; Hasegawa, H.; Wang, F.; Hogan, B.L. The role of Scgb1a1+ Clara cells in the long-term maintenance and repair of lung airway, but not alveolar, epithelium. Cell Stem Cell 2009 , 4 , 525–534. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Travaglini, K.J.; Nabhan, A.N.; Penland, L.; Sinha, R.; Gillich, A.; Sit, R.V.; Chang, S.; Conley, S.D.; Mori, Y.; Seita, J. A molecular cell atlas of the human lung from single-cell RNA sequencing. Nature 2020 , 587 , 619–625. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Vieira Braga, F.A.; Kar, G.; Berg, M.; Carpaij, O.A.; Polanski, K.; Simon, L.M.; Brouwer, S.; Gomes, T.; Hesse, L.; Jiang, J. A cellular census of human lungs identifies novel cell states in health and in asthma. Nat. Med. 2019 , 25 , 1153–1163. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Meyrick, B.; Sturgess, J.M.; Reid, L. A reconstruction of the duct system and secretory tubules of the human bronchial submucosal gland. Thorax 1969 , 24 , 729–736. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Yu, W.; Moninger, T.O.; Thurman, A.L.; Xie, Y.; Jain, A.; Zarei, K.; Powers, L.S.; Pezzulo, A.A.; Stoltz, D.A.; Welsh, M.J. Cellular and molecular architecture of submucosal glands in wild-type and cystic fibrosis pigs. Proc. Natl. Acad. Sci. USA 2022 , 119 , e2119759119. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Desai, T.J.; Brownfield, D.G.; Krasnow, M.A. Alveolar progenitor and stem cells in lung development, renewal and cancer. Nature 2014 , 507 , 190–194. [ Google Scholar ] [ CrossRef ]
- Hogg, J.C.; Macklem, P.T.; Thurlbeck, W. Site and nature of airway obstruction in chronic obstructive lung disease. N. Engl. J. Med. 1968 , 278 , 1355–1360. [ Google Scholar ] [ CrossRef ]
- Hyde, D.M.; Hamid, Q.; Irvin, C.G. Anatomy, pathology, and physiology of the tracheobronchial tree: Emphasis on the distal airways. J. Allergy Clin. Immunol. 2009 , 124 , S72–S77. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Carr, T.F.; Altisheh, R.; Zitt, M. Small airways disease and severe asthma. World Allergy Organ. J. 2017 , 10 , 1–9. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Hogg, J.C.; Paré, P.D.; Hackett, T.-L. The contribution of small airway obstruction to the pathogenesis of chronic obstructive pulmonary disease. Physiol. Rev. 2017 , 97 , 529–552. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Plasschaert, L.W.; Žilionis, R.; Choo-Wing, R.; Savova, V.; Knehr, J.; Roma, G.; Klein, A.M.; Jaffe, A.B. A single-cell atlas of the airway epithelium reveals the CFTR-rich pulmonary ionocyte. Nature 2018 , 560 , 377–381. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Montoro, D.T.; Haber, A.L.; Biton, M.; Vinarsky, V.; Lin, B.; Birket, S.E.; Yuan, F.; Chen, S.; Leung, H.M.; Villoria, J. A revised airway epithelial hierarchy includes CFTR-expressing ionocytes. Nature 2018 , 560 , 319–324. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Scudieri, P.; Musante, I.; Venturini, A.; Guidone, D.; Genovese, M.; Cresta, F.; Caci, E.; Palleschi, A.; Poeta, M.; Santamaria, F.; et al. Ionocytes and CFTR Chloride Channel Expression in Normal and Cystic Fibrosis Nasal and Bronchial Epithelial Cells. Cells 2020 , 9 , 2090. [ Google Scholar ] [ CrossRef ]
- Okuda, K.; Dang, H.; Kobayashi, Y.; Carraro, G.; Nakano, S.; Chen, G.; Kato, T.; Asakura, T.; Gilmore, R.C.; Morton, L.C.; et al. Secretory Cells Dominate Airway CFTR Expression and Function in Human Airway Superficial Epithelia. Am. J. Respir. Crit. Care Med. 2021 , 203 , 1275–1289. [ Google Scholar ] [ CrossRef ]
- McCarron, A.; Donnelley, M.; Parsons, D. Airway disease phenotypes in animal models of cystic fibrosis. Respir. Res. 2018 , 19 , 54. [ Google Scholar ] [ CrossRef ]
- Rosen, B.H.; Chanson, M.; Gawenis, L.R.; Liu, J.; Sofoluwe, A.; Zoso, A.; Engelhardt, J.F. Animal and model systems for studying cystic fibrosis. J. Cyst. Fibros. 2018 , 17 , S28–S34. [ Google Scholar ] [ CrossRef ]
- Yuan, F.; Gasser, G.N.; Lemire, E.; Montoro, D.T.; Jagadeesh, K.; Zhang, Y.; Duan, Y.; Ievlev, V.; Wells, K.L.; Rotti, P.G.; et al. Transgenic ferret models define pulmonary ionocyte diversity and function. Nature 2023 , 621 , 857–867. [ Google Scholar ] [ CrossRef ]
- Sato, Y.; Kim, D.; Turner, M.J.; Luo, Y.; Zaidi, S.S.Z.; Thomas, D.Y.; Hanrahan, J.W. Ionocyte-Specific Regulation of Cystic Fibrosis Transmembrane Conductance Regulator. Am. J. Respir. Cell Mol. Biol. 2023 , 69 , 281–294. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Cai, Q.; Luo, M.; Tang, Y.; Yu, M.; Yuan, F.; Gasser, G.N.; Liu, X.; Engelhardt, J.F. Sonic Hedgehog Signaling Is Essential for Pulmonary Ionocyte Specification in Human and Ferret Airway Epithelia. Am. J. Respir. Cell Mol. Biol. 2023 , 69 , 295–309. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Wang, R.; Simone-Roach, C.; Lindstrom-Vautrin, J.; Wang, F.; Rollins, S.; Bawa, P.S.; Lu, J.; Tang, Y.; Beermann, M.L.; Schlaeger, T.; et al. De Novo Generation of Pulmonary Ionocytes from Normal and Cystic Fibrosis Human Induced Pluripotent Stem Cells. Am. J. Respir. Crit. Care Med. 2023 , 207 , 1249–1253. [ Google Scholar ] [ CrossRef ]
- Jeffery, P.; Li, D. Airway mucosa: Secretory cells, mucus and mucin genes. Eur. Respir. J. 1997 , 10 , 1655–1662. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Roy, M.G.; Livraghi-Butrico, A.; Fletcher, A.A.; McElwee, M.M.; Evans, S.E.; Boerner, R.M.; Alexander, S.N.; Bellinghausen, L.K.; Song, A.S.; Petrova, Y.M.; et al. Muc5b is required for airway defence. Nature 2014 , 505 , 412–416. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Jayaraman, S.; Joo, N.S.; Reitz, B.; Wine, J.J.; Verkman, A.S. Submucosal gland secretions in airways from cystic fibrosis patients have normal [Na(+)] and pH but elevated viscosity. Proc. Natl. Acad. Sci. USA 2001 , 98 , 8119–8123. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Guo, M.; Morley, M.P.; Jiang, C.; Wu, Y.; Li, G.; Du, Y.; Zhao, S.; Wagner, A.; Cakar, A.C.; Kouril, M.; et al. Guided construction of single cell reference for human and mouse lung. Nat. Commun. 2023 , 14 , 4566. [ Google Scholar ] [ CrossRef ]
- Basil, M.C.; Cardenas-Diaz, F.L.; Kathiriya, J.J.; Morley, M.P.; Carl, J.; Brumwell, A.N.; Katzen, J.; Slovik, K.J.; Babu, A.; Zhou, S.; et al. Human distal airways contain a multipotent secretory cell that can regenerate alveoli. Nature 2022 , 604 , 120–126. [ Google Scholar ] [ CrossRef ]
- Carraro, G.; Langerman, J.; Sabri, S.; Lorenzana, Z.; Purkayastha, A.; Zhang, G.; Konda, B.; Aros, C.J.; Calvert, B.A.; Szymaniak, A. Transcriptional analysis of cystic fibrosis airways at single-cell resolution reveals altered epithelial cell states and composition. Nat. Med. 2021 , 27 , 806–814. [ Google Scholar ] [ CrossRef ]
- Carraro, G.; Mulay, A.; Yao, C.; Mizuno, T.; Konda, B.; Petrov, M.; Lafkas, D.; Arron, J.R.; Hogaboam, C.M.; Chen, P.; et al. Single-Cell Reconstruction of Human Basal Cell Diversity in Normal and Idiopathic Pulmonary Fibrosis Lungs. Am. J. Respir. Crit. Care Med. 2020 , 202 , 1540–1550. [ Google Scholar ] [ CrossRef ]
- Beppu, A.K.; Zhao, J.; Yao, C.; Carraro, G.; Israely, E.; Coelho, A.L.; Drake, K.; Hogaboam, C.M.; Parks, W.C.; Kolls, J.K.; et al. Epithelial plasticity and innate immune activation promote lung tissue remodeling following respiratory viral infection. Nat. Commun. 2023 , 14 , 5814. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Sato, Y.; Mustafina, K.R.; Luo, Y.; Martini, C.; Thomas, D.Y.; Wiseman, P.W.; Hanrahan, J.W. Nonspecific binding of common anti-CFTR antibodies in ciliated cells of human airway epithelium. Sci. Rep. 2021 , 11 , 23256. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Xu, Y.; Mizuno, T.; Sridharan, A.; Du, Y.; Guo, M.; Tang, J.; Wikenheiser-Brokamp, K.A.; Perl, A.T.; Funari, V.A.; Gokey, J.J.; et al. Single-cell RNA sequencing identifies diverse roles of epithelial cells in idiopathic pulmonary fibrosis. JCI Insight 2016 , 1 , e90558. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Lotti, V.; Merigo, F.; Lagni, A.; Di Clemente, A.; Ligozzi, M.; Bernardi, P.; Rossini, G.; Concia, E.; Plebani, R.; Romano, M.; et al. CFTR Modulation Reduces SARS-CoV-2 Infection in Human Bronchial Epithelial Cells. Cells 2022 , 11 , 1347. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Lähnemann, D.; Köster, J.; Szczurek, E.; McCarthy, D.J.; Hicks, S.C.; Robinson, M.D.; Vallejos, C.A.; Campbell, K.R.; Beerenwinkel, N.; Mahfouz, A. Eleven grand challenges in single-cell data science. Genome Biol. 2020 , 21 , 31. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Januska, M.N.; Walsh, M.J. Single-Cell RNA Sequencing Reveals New Basic and Translational Insights in the Cystic Fibrosis Lung. Am. J. Respir. Cell Mol. Biol. 2023 , 68 , 131–139. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Schupp, J.C.; Khanal, S.; Gomez, J.L.; Sauler, M.; Adams, T.S.; Chupp, G.L.; Yan, X.; Poli, S.; Zhao, Y.; Montgomery, R.R. Single-cell transcriptional archetypes of airway inflammation in cystic fibrosis. Am. J. Respir. Crit. Care Med. 2020 , 202 , 1419–1429. [ Google Scholar ] [ CrossRef ]
- Leir, S.-H.; Yin, S.; Kerschner, J.L.; Cosme, W.; Harris, A. An atlas of human proximal epididymis reveals cell-specific functions and distinct roles for CFTR. Life Sci. Alliance 2020 , 3 , e202000744. [ Google Scholar ] [ CrossRef ]
- Busslinger, G.A.; Weusten, B.L.; Bogte, A.; Begthel, H.; Brosens, L.A.; Clevers, H. Human gastrointestinal epithelia of the esophagus, stomach, and duodenum resolved at single-cell resolution. Cell Rep. 2021 , 34 , 108819. [ Google Scholar ] [ CrossRef ]
- Burclaff, J.; Bliton, R.J.; Breau, K.A.; Ok, M.T.; Gomez-Martinez, I.; Ranek, J.S.; Bhatt, A.P.; Purvis, J.E.; Woosley, J.T.; Magness, S.T. A proximal-to-distal survey of healthy adult human small intestine and colon epithelium by single-cell transcriptomics. Cell. Mol. Gastroenterol. Hepatol. 2022 , 13 , 1554–1589. [ Google Scholar ] [ CrossRef ]
- Hudson, R.P.; Dawson, J.E.; Chong, P.A.; Yang, Z.; Millen, L.; Thomas, P.J.; Brouillette, C.G.; Forman-Kay, J.D. Direct binding of the corrector VX-809 to human CFTR NBD1: Evidence of an allosteric coupling between the binding site and the NBD1: CL4 interface. Mol. Pharmacol. 2017 , 92 , 124–135. [ Google Scholar ] [ CrossRef ]
- Krainer, G.; Treff, A.; Hartmann, A.; Stone, T.A.; Schenkel, M.; Keller, S.; Deber, C.M.; Schlierf, M. A minimal helical-hairpin motif provides molecular-level insights into misfolding and pharmacological rescue of CFTR. Commun. Biol. 2018 , 1 , 154. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Loo, T.W.; Clarke, D.M. Corrector VX-809 promotes interactions between cytoplasmic loop one and the first nucleotide-binding domain of CFTR. Biochem. Pharmacol. 2017 , 136 , 24–31. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Arora, K.; Moon, C.; Zhang, W.; Yarlagadda, S.; Penmatsa, H.; Ren, A.; Sinha, C.; Naren, A.P. Stabilizing rescued surface-localized δf508 CFTR by potentiation of its interaction with Na(+)/H(+) exchanger regulatory factor 1. Biochemistry 2014 , 53 , 4169–4179. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Pankow, S.; Bamberger, C.; Calzolari, D.; Martínez-Bartolomé, S.; Lavallée-Adam, M.; Balch, W.E.; Yates, J.R., III. ∆F508 CFTR interactome remodelling promotes rescue of cystic fibrosis. Nature 2015 , 528 , 510–516. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Pankow, S.; Bamberger, C.; Yates, J.R., 3rd. A posttranslational modification code for CFTR maturation is altered in cystic fibrosis. Sci. Signal 2019 , 12 , eaan7984. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Gingras, A.C.; Abe, K.T.; Raught, B. Getting to know the neighborhood: Using proximity-dependent biotinylation to characterize protein complexes and map organelles. Curr. Opin. Chem. Biol. 2019 , 48 , 44–54. [ Google Scholar ] [ CrossRef ]
- Cho, K.F.; Branon, T.C.; Rajeev, S.; Svinkina, T.; Udeshi, N.D.; Thoudam, T.; Kwak, C.; Rhee, H.W.; Lee, I.K.; Carr, S.A.; et al. Split-TurboID enables contact-dependent proximity labeling in cells. Proc. Natl. Acad. Sci. USA 2020 , 117 , 12143–12154. [ Google Scholar ] [ CrossRef ]
- Branon, T.C.; Bosch, J.A.; Sanchez, A.D.; Udeshi, N.D.; Svinkina, T.; Carr, S.A.; Feldman, J.L.; Perrimon, N.; Ting, A.Y. Efficient proximity labeling in living cells and organisms with TurboID. Nat. Biotechnol. 2018 , 36 , 880–887. [ Google Scholar ] [ CrossRef ]
- Chevalier, B.; Baatallah, N.; Najm, M.; Castanier, S.; Jung, V.; Pranke, I.; Golec, A.; Stoven, V.; Marullo, S.; Antigny, F.; et al. Differential CFTR-Interactome Proximity Labeling Procedures Identify Enrichment in Multiple SLC Transporters. Int. J. Mol. Sci. 2022 , 23 , 8937. [ Google Scholar ] [ CrossRef ]
- Hutt, D.M.; Loguercio, S.; Campos, A.R.; Balch, W.E. A proteomic variant approach (ProVarA) for personalized medicine of inherited and somatic disease. J. Mol. Biol. 2018 , 430 , 2951–2973. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Sood, R.; Bear, C.; Auerbach, W.; Reyes, E.; Jensen, T.; Kartner, N.; Riordan, J.R.; Buchwald, M. Regulation of CFTR expression and function during differentiation of intestinal epithelial cells. EMBO J. 1992 , 11 , 2487–2494. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Monterisi, S.; Favia, M.; Guerra, L.; Cardone, R.A.; Marzulli, D.; Reshkin, S.J.; Casavola, V.; Zaccolo, M. CFTR regulation in human airway epithelial cells requires integrity of the actin cytoskeleton and compartmentalized cAMP and PKA activity. J. Cell Sci. 2012 , 125 Pt 5 , 1106–1117. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Edelman, A. Cytoskeleton and CFTR. Int. J. Biochem. Cell Biol. 2014 , 52 , 68–72. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Wang, X.; Venable, J.; LaPointe, P.; Hutt, D.M.; Koulov, A.V.; Coppinger, J.; Gurkan, C.; Kellner, W.; Matteson, J.; Plutner, H.; et al. Hsp90 cochaperone Aha1 downregulation rescues misfolding of CFTR in cystic fibrosis. Cell 2006 , 127 , 803–815. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Chanoux, R.A.; Rubenstein, R.C. Molecular Chaperones as Targets to Circumvent the CFTR Defect in Cystic Fibrosis. Front. Pharmacol. 2012 , 3 , 137. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Ameen, N.; Silvis, M.; Bradbury, N.A. Endocytic trafficking of CFTR in health and disease. J. Cyst. Fibros. 2007 , 6 , 1–14. [ Google Scholar ] [ CrossRef ]
- Sun, F.; Hug, M.J.; Bradbury, N.A.; Frizzell, R.A. Protein kinase A associates with cystic fibrosis transmembrane conductance regulator via an interaction with ezrin. J. Biol. Chem. 2000 , 275 , 14360–14366. [ Google Scholar ] [ CrossRef ]
- Klein, H.; Abu-Arish, A.; Trinh, N.T.; Luo, Y.; Wiseman, P.W.; Hanrahan, J.W.; Brochiero, E.; Sauvé, R. Investigating CFTR and KCa3.1 Protein/Protein Interactions. PLoS ONE 2016 , 11 , e0153665. [ Google Scholar ] [ CrossRef ]
- Bertrand, C.A.; Mitra, S.; Mishra, S.K.; Wang, X.; Zhao, Y.; Pilewski, J.M.; Madden, D.R.; Frizzell, R.A. The CFTR trafficking mutation F508del inhibits the constitutive activity of SLC26A9. Am. J. Physiol. Lung Cell Mol. Physiol. 2017 , 312 , L912–L925. [ Google Scholar ] [ CrossRef ]
- Davezac, N.; Tondelier, D.; Lipecka, J.; Fanen, P.; Demaugre, F.; Debski, J.; Dadlez, M.; Schrattenholz, A.; Cahill, M.A.; Edelman, A. Global proteomic approach unmasks involvement of keratins 8 and 18 in the delivery of cystic fibrosis transmembrane conductance regulator (CFTR)/ΔF508-CFTR to the plasma membrane. Proteomics 2004 , 4 , 3833–3844. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Ramalho, S.S.; Silva, I.A.; Amaral, M.D.; Farinha, C.M. Rare trafficking CFTR mutations involve distinct cellular retention machineries and require different rescuing strategies. Int. J. Mol. Sci. 2021 , 23 , 24. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Lu, Y.; Xiong, X.; Helm, A.; Kimani, K.; Bragin, A.; Skach, W.R. Co-and posttranslational translocation mechanisms direct cystic fibrosis transmembrane conductance regulator N terminus transmembrane assembly. J. Biol. Chem. 1998 , 273 , 568–576. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Chen, M.; Zhang, J.T. Membrane insertion, processing, and topology of cystic fibrosis transmembrane conductance regulator (CFTR) in microsomal membranes. Mol. Membr. Biol. 1996 , 13 , 33–40. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Pitonzo, D.; Yang, Z.; Matsumura, Y.; Johnson, A.E.; Skach, W.R. Sequence-specific retention and regulated integration of a nascent membrane protein by the endoplasmic reticulum Sec61 translocon. Mol. Biol. Cell 2009 , 20 , 685–698. [ Google Scholar ] [ CrossRef ]
- Yang, Y.; Janich, S.; Cohn, J.A.; Wilson, J.M. The common variant of cystic fibrosis transmembrane conductance regulator is recognized by hsp70 and degraded in a pre-Golgi nonlysosomal compartment. Proc. Natl. Acad. Sci. USA 1993 , 90 , 9480–9484. [ Google Scholar ] [ CrossRef ]
- Kleizen, B.; van Vlijmen, T.; de Jonge, H.R.; Braakman, I. Folding of CFTR is predominantly cotranslational. Mol. Cell 2005 , 20 , 277–287. [ Google Scholar ] [ CrossRef ]
- Lukacs, G.L.; Mohamed, A.; Kartner, N.; Chang, X.B.; Riordan, J.R.; Grinstein, S. Conformational maturation of CFTR but not its mutant counterpart (delta F508) occurs in the endoplasmic reticulum and requires ATP. EMBO J. 1994 , 13 , 6076–6086. [ Google Scholar ] [ CrossRef ]
- Pranke, I.M.; Sermet-Gaudelus, I. Biosynthesis of cystic fibrosis transmembrane conductance regulator. Int. J. Biochem. Cell Biol. 2014 , 52 , 26–38. [ Google Scholar ] [ CrossRef ]
- Estabrooks, S.; Brodsky, J.L. Regulation of CFTR biogenesis by the proteostatic network and pharmacological modulators. Int. J. Mol. Sci. 2020 , 21 , 452. [ Google Scholar ] [ CrossRef ]
- Farinha, C.M.; Canato, S. From the endoplasmic reticulum to the plasma membrane: Mechanisms of CFTR folding and trafficking. Cell. Mol. Life Sci. 2017 , 74 , 39–55. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Coe, H.; Michalak, M. Calcium binding chaperones of the endoplasmic reticulum. Gen. Physiol. Biophys. 2009 , 28 , F96–F103. [ Google Scholar ] [ PubMed ]
- Loo, M.A.; Jensen, T.J.; Cui, L.; Hou, Y.-X.; Chang, X.-B.; Riordan, J.R. Perturbation of Hsp90 interaction with nascent CFTR prevents its maturation and accelerates its degradation by the proteasome. EMBO J. 1998 , 17 , 6879–6887. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Matsumura, Y.; David, L.L.; Skach, W.R. Role of Hsc70 binding cycle in CFTR folding and endoplasmic reticulum–associated degradation. Mol. Biol. Cell 2011 , 22 , 2797–2809. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Meacham, G.C.; Lu, Z.; King, S.; Sorscher, E.; Tousson, A.; Cyr, D.M. The Hdj-2/Hsc70 chaperone pair facilitates early steps in CFTR biogenesis. EMBO J. 1999 , 18 , 1492–1505. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Alberti, S.; Bohse, K.; Arndt, V.; Schmitz, A.; Hohfeld, J. The cochaperone HspBP1 inhibits the CHIP ubiquitin ligase and stimulates the maturation of the cystic fibrosis transmembrane conductance regulator. Mol. Biol. Cell 2004 , 15 , 4003–4010. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Grove, D.E.; Fan, C.-Y.; Ren, H.Y.; Cyr, D.M. The endoplasmic reticulum–associated Hsp40 DNAJB12 and Hsc70 cooperate to facilitate RMA1 E3–dependent degradation of nascent CFTRΔF508. Mol. Biol. Cell 2011 , 22 , 301–314. [ Google Scholar ] [ CrossRef ]
- Yamamoto, Y.-h.; Kimura, T.; Momohara, S.; Takeuchi, M.; Tani, T.; Kimata, Y.; Kadokura, H.; Kohno, K. A novel ER J-protein DNAJB12 accelerates ER-associated degradation of membrane proteins including CFTR. Cell Struct. Funct. 2010 , 35 , 107–116. [ Google Scholar ] [ CrossRef ]
- Farinha, C.M.; Nogueira, P.; Mendes, F.; Penque, D.; Amaral, M.D. The human DnaJ homologue (Hdj)-1/heat-shock protein (Hsp) 40 co-chaperone is required for the in vivo stabilization of the cystic fibrosis transmembrane conductance regulator by Hsp70. Biochem. J. 2002 , 366 , 797–806. [ Google Scholar ] [ CrossRef ]
- Zhang, H.; Schmidt, B.Z.; Sun, F.; Condliffe, S.B.; Butterworth, M.B.; Youker, R.T.; Brodsky, J.L.; Aridor, M.; Frizzell, R.A. Cysteine string protein monitors late steps in cystic fibrosis transmembrane conductance regulator biogenesis. J. Biol. Chem. 2006 , 281 , 11312–11321. [ Google Scholar ] [ CrossRef ]
- Huang, Y.; Arora, K.; Mun, K.S.; Yang, F.; Moon, C.; Yarlagadda, S.; Jegga, A.; Weaver, T.; Naren, A.P. Targeting DNAJB9, a novel ER luminal co-chaperone, to rescue ΔF508-CFTR. Sci. Rep. 2019 , 9 , 9808. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Sharma, M.; Pampinella, F.; Nemes, C.; Benharouga, M.; So, J.; Du, K.; Bache, K.G.; Papsin, B.; Zerangue, N.; Stenmark, H. Misfolding diverts CFTR from recycling to degradation: Quality control at early endosomes. J. Cell Biol. 2004 , 164 , 923–933. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Weixel, K.M.; Bradbury, N.A. The carboxyl terminus of the cystic fibrosis transmembrane conductance regulator binds to AP-2 clathrin adaptors. J. Biol. Chem. 2000 , 275 , 3655–3660. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Fu, L.; Rab, A.; Tang, L.P.; Rowe, S.M.; Bebok, Z.; Collawn, J.F. Dab2 is a key regulator of endocytosis and post-endocytic trafficking of the cystic fibrosis transmembrane conductance regulator. Biochem. J. 2012 , 441 , 633–643. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Gentzsch, M.; Chang, X.-B.; Cui, L.; Wu, Y.; Ozols, V.V.; Choudhury, A.; Pagano, R.E.; Riordan, J.R. Endocytic trafficking routes of wild type and ΔF508 cystic fibrosis transmembrane conductance regulator. Mol. Biol. Cell 2004 , 15 , 2684–2696. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Cheng, J.; Moyer, B.D.; Milewski, M.; Loffing, J.; Ikeda, M.; Mickle, J.E.; Cutting, G.R.; Li, M.; Stanton, B.A.; Guggino, W.B. A Golgi-associated PDZ domain protein modulates cystic fibrosis transmembrane regulator plasma membrane expression. J. Biol. Chem. 2002 , 277 , 3520–3529. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Peters, K.W.; Qi, J.; Watkins, S.C.; Frizzell, R.A. Syntaxin 1A inhibits regulated CFTR trafficking in Xenopus oocytes . Am. J. Physiol. Cell Physiol. 1999 , 277 , C174–C180. [ Google Scholar ] [ CrossRef ]
- Arora, K.; Liyanage, P.; Zhong, Q.; Naren, A.P. A SNARE protein Syntaxin 17 captures CFTR to potentiate autophagosomal clearance under stress. FASEB J. 2021 , 35 , e21185. [ Google Scholar ] [ CrossRef ]
- Assani, K.; Tazi, M.F.; Amer, A.O.; Kopp, B.T. IFN-γ stimulates autophagy-mediated clearance of Burkholderia cenocepacia in human cystic fibrosis macrophages. PLoS ONE 2014 , 9 , e96681. [ Google Scholar ] [ CrossRef ]
- Yuan, K.; Huang, C.; Fox, J.; Laturnus, D.; Carlson, E.; Zhang, B.; Yin, Q.; Gao, H.; Wu, M. Autophagy plays an essential role in the clearance of Pseudomonas aeruginosa by alveolar macrophages. J. Cell Sci. 2012 , 125 , 507–515. [ Google Scholar ] [ CrossRef ]
- Della Sala, A.; Prono, G.; Hirsch, E.; Ghigo, A. Role of protein kinase A-mediated phosphorylation in CFTR channel activity regulation. Front. Physiol. 2021 , 12 , 690247. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Chin, S.; Hung, M.; Bear, C.E. Current insights into the role of PKA phosphorylation in CFTR channel activity and the pharmacological rescue of cystic fibrosis disease-causing mutants. Cell. Mol. Life Sci. 2017 , 74 , 57–66. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Seavilleklein, G.; Amer, N.; Evagelidis, A.; Chappe, F.; Irvine, T.; Hanrahan, J.W.; Chappe, V. PKC phosphorylation modulates PKA-dependent binding of the R domain to other domains of CFTR. Am. J. Physiol. Cell Physiol. 2008 , 295 , C1366–C1375. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Chappe, V.; Hinkson, D.A.; Howell, L.D.; Evagelidis, A.; Liao, J.; Chang, X.-B.; Riordan, J.R.; Hanrahan, J.W. Stimulatory and inhibitory protein kinase C consensus sequences regulate the cystic fibrosis transmembrane conductance regulator. Proc. Natl. Acad. Sci. USA 2004 , 101 , 390–395. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Billet, A.; Luo, Y.; Balghi, H.; Hanrahan, J.W. Role of tyrosine phosphorylation in the muscarinic activation of the cystic fibrosis transmembrane conductance regulator (CFTR). J. Biol. Chem. 2013 , 288 , 21815–21823. [ Google Scholar ] [ CrossRef ]
- Ren, A.; Zhang, W.; Yarlagadda, S.; Sinha, C.; Arora, K.; Moon, C.S.; Naren, A.P. MAST205 competes with cystic fibrosis transmembrane conductance regulator (CFTR)-associated ligand for binding to CFTR to regulate CFTR-mediated fluid transport. J. Biol. Chem. 2013 , 288 , 12325–12334. [ Google Scholar ] [ CrossRef ]
- Zhang, W.; Zhang, Z.; Zhang, Y.; Naren, A.P. CFTR-NHERF2-LPA2 complex in the airway and gut epithelia. Int. J. Mol. Sci. 2017 , 18 , 1896. [ Google Scholar ] [ CrossRef ]
- Naren, A.P.; Cobb, B.; Li, C.; Roy, K.; Nelson, D.; Heda, G.D.; Liao, J.; Kirk, K.L.; Sorscher, E.J.; Hanrahan, J. A macromolecular complex of β2 adrenergic receptor, CFTR, and ezrin/radixin/moesin-binding phosphoprotein 50 is regulated by PKA. Proc. Natl. Acad. Sci. USA 2003 , 100 , 342–346. [ Google Scholar ] [ CrossRef ]
- Li, C.; Dandridge, K.S.; Di, A.; Marrs, K.L.; Harris, E.L.; Roy, K.; Jackson, J.S.; Makarova, N.V.; Fujiwara, Y.; Farrar, P.L. Lysophosphatidic acid inhibits cholera toxin-induced secretory diarrhea through CFTR-dependent protein interactions. J. Exp. Med. 2005 , 202 , 975–986. [ Google Scholar ] [ CrossRef ]
- Li, C.; Krishnamurthy, P.C.; Penmatsa, H.; Marrs, K.L.; Wang, X.Q.; Zaccolo, M.; Jalink, K.; Li, M.; Nelson, D.J.; Schuetz, J.D. Spatiotemporal coupling of cAMP transporter to CFTR chloride channel function in the gut epithelia. Cell 2007 , 131 , 940–951. [ Google Scholar ] [ CrossRef ]
- Arora, K.; Sinha, C.; Zhang, W.; Moon, C.S.; Ren, A.; Yarlagadda, S.; Dostmann, W.R.; Adebiyi, A.; Haberman, Y.; Denson, L.A. Altered cGMP dynamics at the plasma membrane contribute to diarrhea in ulcerative colitis. Am. J. Pathol. 2015 , 185 , 2790–2804. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Sun, F.; Hug, M.J.; Lewarchik, C.M.; Yun, C.-H.C.; Bradbury, N.A.; Frizzell, R.A. E3KARP mediates the association of ezrin and protein kinase A with the cystic fibrosis transmembrane conductance regulator in airway cells. J. Biol. Chem. 2000 , 275 , 29539–29546. [ Google Scholar ] [ CrossRef ]
- Sadana, R.; Dessauer, C.W. Physiological roles for G protein-regulated adenylyl cyclase isoforms: Insights from knockout and overexpression studies. Neurosignals 2009 , 17 , 5–22. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Frizzell, R.A.; Hanrahan, J.W. Physiology of epithelial chloride and fluid secretion. Cold Spring Harb. Perspect. Med. 2012 , 2 , a009563. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Field, M. Intestinal secretion: Effect of cyclic AMP and its role in cholera. N. Engl. J. Med. 1971 , 284 , 1137–1144. [ Google Scholar ]
- Bartlett, S. Water, sanitation and urban children: The need to go beyond “improved” provision. Environ. Urban. 2003 , 15 , 57–70. [ Google Scholar ]
- Demissie, G.D.; Yeshaw, Y.; Aleminew, W.; Akalu, Y. Diarrhea and associated factors among under five children in sub-Saharan Africa: Evidence from demographic and health surveys of 34 sub-Saharan countries. PLoS ONE 2021 , 16 , e0257522. [ Google Scholar ] [ CrossRef ]
- Kimberg, D.V.; Field, M.; Johnson, J.; Henderson, A.; Gershon, E. Stimulation of intestinal mucosal adenyl cyclase by cholera enterotoxin and prostaglandins. J. Clin. Investig. 1971 , 50 , 1218–1230. [ Google Scholar ] [ CrossRef ]
- Kantor, H.S. Enterotoxins of Escherichia coli and Vibrio cholerae: Tools for the molecular biologist. J. Infect. Dis. 1975 , 131 , s22–s32. [ Google Scholar ] [ CrossRef ]
- Vaandrager, A.B.; Bot, A.G.; Ruth, P.; Pfeifer, A.; Hofmann, F.; De Jonge, H.R. Differential role of cyclic GMP–dependent protein kinase II in ion transport in murine small intestine and colon. Gastroenterology 2000 , 118 , 108–114. [ Google Scholar ] [ CrossRef ]
- Vaandrager, A.B.; Smolenski, A.; Tilly, B.C.; Houtsmuller, A.B.; Ehlert, E.M.; Bot, A.G.; Edixhoven, M.; Boomaars, W.E.; Lohmann, S.M.; De Jonge, H.R. Membrane targeting of cGMP-dependent protein kinase is required for cystic fibrosis transmembrane conductance regulator Cl − channel activation. Proc. Natl. Acad. Sci. USA 1998 , 95 , 1466–1471. [ Google Scholar ] [ CrossRef ]
- Francis, S.H.; Turko, I.V.; Corbin, J.D. Cyclic nucleotide phosphodiesterases: Relating structure and function. Prog. Nucleic Acid. Res. Mol. Biol. 2000 , 65 , 1–52. [ Google Scholar ]
- Penmatsa, H.; Zhang, W.; Yarlagadda, S.; Li, C.; Conoley, V.G.; Yue, J.; Bahouth, S.W.; Buddington, R.K.; Zhang, G.; Nelson, D.J. Compartmentalized cyclic adenosine 3′,5′-monophosphate at the plasma membrane clusters PDE3A and cystic fibrosis transmembrane conductance regulator into microdomains. Mol. Biol. Cell 2010 , 21 , 1097–1110. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Thomas, A.; Ramananda, Y.; Mun, K.; Naren, A.P.; Arora, K. AC6 is the major adenylate cyclase forming a diarrheagenic protein complex with cystic fibrosis transmembrane conductance regulator in cholera. J. Biol. Chem. 2018 , 293 , 12949–12959. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Fenton, R.A.; Murali, S.K.; Kaji, I.; Akiba, Y.; Kaunitz, J.D.; Kristensen, T.B.; Poulsen, S.B.; Dominguez Rieg, J.A.; Rieg, T. Adenylyl Cyclase 6 Expression Is Essential for Cholera Toxin-Induced Diarrhea. J. Infect. Dis. 2019 , 220 , 1719–1728. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Arora, K.; Sinha, C.; Zhang, W.; Ren, A.; Moon, C.S.; Yarlagadda, S.; Naren, A.P. Compartmentalization of cyclic nucleotide signaling: A question of when, where, and why? Pflügers Arch. Eur. J. Physiol. 2013 , 465 , 1397–1407. [ Google Scholar ] [ CrossRef ] [ PubMed ]
- Arora, K.; Lund, J.R.; Naren, N.A.; Zingarelli, B.; Naren, A.P. AC6 regulates the microtubule-depolymerizing kinesin KIF19A to control ciliary length in mammals. J. Biol. Chem. 2020 , 295 , 14250–14259. [ Google Scholar ] [ CrossRef ]
- Basu, N.; Arshad, N.; Visweswariah, S.S. Receptor guanylyl cyclase C (GC-C): Regulation and signal transduction. Mol. Cell. Biochem. 2010 , 334 , 67–80. [ Google Scholar ] [ CrossRef ]
- Navaneethan, U.; Giannella, R.A. Mechanisms of infectious diarrhea. Nat. Clin. Pract. Gastroenterol. Hepatol. 2008 , 5 , 637–647. [ Google Scholar ] [ CrossRef ]
- Schulz, S.; Green, C.K.; Yuen, P.S.; Garbers, D.L. Guanylyl cyclase is a heat-stable enterotoxin receptor. Cell 1990 , 63 , 941–948. [ Google Scholar ] [ CrossRef ]
- Fiskerstrand, T.; Arshad, N.; Haukanes, B.I.; Tronstad, R.R.; Pham, K.D.-C.; Johansson, S.; Håvik, B.; Tønder, S.L.; Levy, S.E.; Brackman, D. Familial diarrhea syndrome caused by an activating GUCY2C mutation. N. Engl. J. Med. 2012 , 366 , 1586–1595. [ Google Scholar ] [ CrossRef ]
- Romi, H.; Cohen, I.; Landau, D.; Alkrinawi, S.; Yerushalmi, B.; Hershkovitz, R.; Newman-Heiman, N.; Cutting, G.R.; Ofir, R.; Sivan, S. Meconium ileus caused by mutations in GUCY2C, encoding the CFTR-activating guanylate cyclase 2C. Am. J. Hum. Genet. 2012 , 90 , 893–899. [ Google Scholar ] [ CrossRef ]
- Corsetti, M.; Tack, J. Linaclotide: A new drug for the treatment of chronic constipation and irritable bowel syndrome with constipation. United Eur. Gastroenterol. J. 2013 , 1 , 7–20. [ Google Scholar ] [ CrossRef ]
- Arora, K.; Huang, Y.; Mun, K.; Yarlagadda, S.; Sundaram, N.; Kessler, M.M.; Hannig, G.; Kurtz, C.B.; Silos-Santiago, I.; Helmrath, M.; et al. Guanylate cyclase 2C agonism corrects CFTR mutants. JCI Insight 2017 , 2 , 93686. [ Google Scholar ] [ CrossRef ]
Click here to enlarge figure
| The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
Share and Cite
Ramananda, Y.; Naren, A.P.; Arora, K. Functional Consequences of CFTR Interactions in Cystic Fibrosis. Int. J. Mol. Sci. 2024 , 25 , 3384. https://doi.org/10.3390/ijms25063384
Ramananda Y, Naren AP, Arora K. Functional Consequences of CFTR Interactions in Cystic Fibrosis. International Journal of Molecular Sciences . 2024; 25(6):3384. https://doi.org/10.3390/ijms25063384
Ramananda, Yashaswini, Anjaparavanda P. Naren, and Kavisha Arora. 2024. "Functional Consequences of CFTR Interactions in Cystic Fibrosis" International Journal of Molecular Sciences 25, no. 6: 3384. https://doi.org/10.3390/ijms25063384
Article Metrics
Article access statistics, further information, mdpi initiatives, follow mdpi.
Subscribe to receive issue release notifications and newsletters from MDPI journals

An official website of the United States government
The .gov means it’s official. Federal government websites often end in .gov or .mil. Before sharing sensitive information, make sure you’re on a federal government site.
The site is secure. The https:// ensures that you are connecting to the official website and that any information you provide is encrypted and transmitted securely.
- Publications
- Account settings
- My Bibliography
- Collections
- Citation manager
Save citation to file
Email citation, add to collections.
- Create a new collection
- Add to an existing collection
Add to My Bibliography
Your saved search, create a file for external citation management software, your rss feed.
- Search in PubMed
- Search in NLM Catalog
- Add to Search
Cystic Fibrosis Transmembrane Conductance Regulator (CFTR): CLOSED AND OPEN STATE CHANNEL MODELS
Affiliations.
- 1 From the Department of Biological Sciences and Centre for Molecular Simulation, University of Calgary, Calgary, Alberta T2N 1N4, Canada and.
- 2 Research Department of Neuroscience, Physiology and Pharmacology, University College London, Gower Street, London WC1E 6BT, United Kingdom [email protected].
- 3 From the Department of Biological Sciences and Centre for Molecular Simulation, University of Calgary, Calgary, Alberta T2N 1N4, Canada and [email protected].
- PMID: 26229102
- PMCID: PMC4645605
- DOI: 10.1074/jbc.M115.665125
The cystic fibrosis transmembrane conductance regulator (CFTR) is a member of the ATP-binding cassette (ABC) transporter superfamily. CFTR controls the flow of anions through the apical membrane of epithelia. Dysfunctional CFTR causes the common lethal genetic disease cystic fibrosis. Transitions between open and closed states of CFTR are regulated by ATP binding and hydrolysis on the cytosolic nucleotide binding domains, which are coupled with the transmembrane (TM) domains forming the pathway for anion permeation. Lack of structural data hampers a global understanding of CFTR and thus the development of "rational" approaches directly targeting defective CFTR. In this work, we explored possible conformational states of the CFTR gating cycle by means of homology modeling. As templates, we used structures of homologous ABC transporters, namely TM(287-288), ABC-B10, McjD, and Sav1866. In the light of published experimental results, structural analysis of the transmembrane cavity suggests that the TM(287-288)-based CFTR model could correspond to a commonly occupied closed state, whereas the McjD-based model could represent an open state. The models capture the important role played by Phe-337 as a filter/gating residue and provide structural information on the conformational transition from closed to open channel.
Keywords: ABC transporter; chloride transport; cystic fibrosis; cystic fibrosis transmembrane conductance regulator (CFTR); molecular modeling.
© 2015 by The American Society for Biochemistry and Molecular Biology, Inc.
PubMed Disclaimer
Similar articles
- Conserved allosteric hot spots in the transmembrane domains of cystic fibrosis transmembrane conductance regulator (CFTR) channels and multidrug resistance protein (MRP) pumps. Wei S, Roessler BC, Chauvet S, Guo J, Hartman JL 4th, Kirk KL. Wei S, et al. J Biol Chem. 2014 Jul 18;289(29):19942-57. doi: 10.1074/jbc.M114.562116. Epub 2014 May 29. J Biol Chem. 2014. PMID: 24876383 Free PMC article.
- Evolutionary and functional divergence between the cystic fibrosis transmembrane conductance regulator and related ATP-binding cassette transporters. Jordan IK, Kota KC, Cui G, Thompson CH, McCarty NA. Jordan IK, et al. Proc Natl Acad Sci U S A. 2008 Dec 2;105(48):18865-70. doi: 10.1073/pnas.0806306105. Epub 2008 Nov 19. Proc Natl Acad Sci U S A. 2008. PMID: 19020075 Free PMC article.
- The gating of the CFTR channel. Moran O. Moran O. Cell Mol Life Sci. 2017 Jan;74(1):85-92. doi: 10.1007/s00018-016-2390-z. Epub 2016 Oct 1. Cell Mol Life Sci. 2017. PMID: 27696113 Free PMC article. Review.
- Metal bridges illuminate transmembrane domain movements during gating of the cystic fibrosis transmembrane conductance regulator chloride channel. El Hiani Y, Linsdell P. El Hiani Y, et al. J Biol Chem. 2014 Oct 10;289(41):28149-59. doi: 10.1074/jbc.M114.593103. Epub 2014 Aug 20. J Biol Chem. 2014. PMID: 25143385 Free PMC article.
- Cystic fibrosis transmembrane conductance regulator (CFTR): Making an ion channel out of an active transporter structure. Linsdell P. Linsdell P. Channels (Austin). 2018;12(1):284-290. doi: 10.1080/19336950.2018.1502585. Channels (Austin). 2018. PMID: 30152709 Free PMC article. Review.
- The burden of cystic fibrosis in North Africa. El Makhzen N, Daimi H, Bouguenouch L, Abriel H. El Makhzen N, et al. Front Genet. 2024 Jan 10;14:1295008. doi: 10.3389/fgene.2023.1295008. eCollection 2023. Front Genet. 2024. PMID: 38269366 Free PMC article. Review.
- ATP-dependent transporters: emerging players at the crossroads of immunity and metabolism. Balasubramanian A, Sundrud MS. Balasubramanian A, et al. Front Immunol. 2023 Oct 31;14:1286696. doi: 10.3389/fimmu.2023.1286696. eCollection 2023. Front Immunol. 2023. PMID: 38022644 Free PMC article. Review.
- Oxidative Stress Biomarkers in Cystic Fibrosis and Cystic Fibrosis-Related Diabetes in Children: A Literature Review. Pinzaru AD, Mihai CM, Chisnoiu T, Pantazi AC, Lupu VV, Kassim MAK, Lupu A, Grosan E, Al Jumaili AZN, Ion I, Stoleriu G, Ion I. Pinzaru AD, et al. Biomedicines. 2023 Sep 29;11(10):2671. doi: 10.3390/biomedicines11102671. Biomedicines. 2023. PMID: 37893045 Free PMC article. Review.
- Molecular dynamics study of Cl - permeation through cystic fibrosis transmembrane conductance regulator (CFTR). Zeng ZW, Linsdell P, Pomès R. Zeng ZW, et al. Cell Mol Life Sci. 2023 Jan 24;80(2):51. doi: 10.1007/s00018-022-04621-7. Cell Mol Life Sci. 2023. PMID: 36694009 Free PMC article.
- The Distribution and Role of the CFTR Protein in the Intracellular Compartments. Lukasiak A, Zajac M. Lukasiak A, et al. Membranes (Basel). 2021 Oct 22;11(11):804. doi: 10.3390/membranes11110804. Membranes (Basel). 2021. PMID: 34832033 Free PMC article. Review.
- Rommens J. M., Iannuzzi M. C., Kerem B., Drumm M. L., Melmer G., Dean M., Rozmahel R., Cole J. L., Kennedy D., Hidaka N., et al. (1989) Identification of the cystic fibrosis gene: chromosome walking and jumping. Science 245, 1059–1065 - PubMed
- Cheng S. H., Gregory R. J., Marshall J., Paul S., Souza D. W., White G. A., O'Riordan C. R., Smith A. E. (1990) Defective intracellular transport and processing of CFTR is the molecular basis of most cystic fibrosis. Cell 63, 827–834 - PubMed
- Dalemans W., Barbry P., Champigny G., Jallat S., Dott K., Dreyer D., Crystal R. G., Pavirani A., Lecocq J. P., Lazdunski M. (1991) Altered chloride ion channel kinetics associated with the ΔF508 cystic fibrosis mutation. Nature 354, 526–528 - PubMed
- Gadsby D. C., Vergani P., Csanády L. (2006) The ABC protein turned chloride channel whose failure causes cystic fibrosis. Nature 440, 477–483 - PMC - PubMed
- Riordan J. R. (2005) Assembly of functional CFTR chloride channels. Annu. Rev. Physiol. 67, 701–718 - PubMed
Publication types
- Search in MeSH
Associated data
- Search in Structure
Related information
- Gene (GeneRIF)
- Nucleotide (RefSeq)
- Protein (RefSeq)
Grants and funding
- G0501200/MRC_/Medical Research Council/United Kingdom
- Canadian Institutes of Health Research/Canada
LinkOut - more resources
Full text sources.
- Elsevier Science
- Europe PubMed Central
- PubMed Central
- Genetic Alliance
Research Materials
- NCI CPTC Antibody Characterization Program

- Citation Manager
NCBI Literature Resources
MeSH PMC Bookshelf Disclaimer
The PubMed wordmark and PubMed logo are registered trademarks of the U.S. Department of Health and Human Services (HHS). Unauthorized use of these marks is strictly prohibited.
Thank you for visiting nature.com. You are using a browser version with limited support for CSS. To obtain the best experience, we recommend you use a more up to date browser (or turn off compatibility mode in Internet Explorer). In the meantime, to ensure continued support, we are displaying the site without styles and JavaScript.
- View all journals
- Explore content
- About the journal
- Publish with us
- Sign up for alerts
- Published: 14 May 2015
Cystic fibrosis
- Felix Ratjen 1 ,
- Scott C. Bell 2 ,
- Steven M. Rowe 3 ,
- Christopher H. Goss 4 ,
- Alexandra L. Quittner 5 &
- Andrew Bush 6
Nature Reviews Disease Primers volume 1 , Article number: 15010 ( 2015 ) Cite this article
14k Accesses
374 Citations
61 Altmetric
Metrics details
- Genetic testing
- Metabolic disorders
- Respiratory tract diseases
Cystic fibrosis is an autosomal recessive, monogenetic disorder caused by mutations in the cystic fibrosis transmembrane conductance regulator ( CFTR ) gene. The gene defect was first described 25 years ago and much progress has been made since then in our understanding of how CFTR mutations cause disease and how this can be addressed therapeutically. CFTR is a transmembrane protein that transports ions across the surface of epithelial cells. CFTR dysfunction affects many organs; however, lung disease is responsible for the vast majority of morbidity and mortality in patients with cystic fibrosis. Prenatal diagnostics, newborn screening and new treatment algorithms are changing the incidence and the prevalence of the disease. Until recently, the standard of care in cystic fibrosis treatment focused on preventing and treating complications of the disease; now, novel treatment strategies directly targeting the ion channel abnormality are becoming available and it will be important to evaluate how these treatments affect disease progression and the quality of life of patients. In this Primer, we summarize the current knowledge, and provide an outlook on how cystic fibrosis clinical care and research will be affected by new knowledge and therapeutic options in the near future. For an illustrated summary of this Primer, visit: http://go.nature.com/4VrefN
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 1 digital issues and online access to articles
92,52 € per year
only 92,52 € per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout
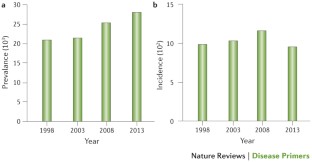
Similar content being viewed by others

Future therapies for cystic fibrosis

CyFi-MAP: an interactive pathway-based resource for cystic fibrosis
Weiler, C. A. & Drumm, M. L. Genetic influences on cystic fibrosis lung disease severity. Front. Pharmacol. 4 , 40 (2013).
Article PubMed PubMed Central Google Scholar
Jarvi, K. et al . Cystic fibrosis transmembrane conductance regulator and obstructive azoospermia. Lancet 345 , 1578 (1995).
Article CAS PubMed Google Scholar
Gonska, T. et al . Role of cystic fibrosis transmembrane conductance regulator in patients with chronic sinopulmonary disease. Chest 142 , 996–1004 (2012).
Wilschanski, M. et al . Mutations in the cystic fibrosis transmembrane regulator gene and in vivo transepithelial potentials. Am. J. Respir. Crit. Care Med. 174 , 787–794 (2006).
Article CAS PubMed PubMed Central Google Scholar
Riordan, J. R. et al . Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science 245 , 1066–1073 (1989).
Rommens, J. M. et al . Identification of the cystic fibrosis gene: chromosome walking and jumping. Science 245 , 1059–1065 (1989).
Ratjen, F. & Grasemann, H. New therapies in cystic fibrosis. Curr. Pharm. Des. 18 , 614–627 (2012).
O'Sullivan, B. P. & Freedman, S. D. Cystic fibrosis. Lancet 373 , 1891–1904 (2009).
Article PubMed Google Scholar
Walters, S., M. A . in Hodson and Geddes' Cystic Fibrosis 3rd edn (eds Hodson, M. E., Geddes D. M., Bush, A. ) 21–45 (Edward Arnold Ltd, 2007).
Google Scholar
Farrell, P. M. The prevalence of cystic fibrosis in the European Union. J. Cyst. Fibros. 7 , 450–453 (2008).
Festini, F. et al . Incidence of cystic fibrosis in the Albanian population. Pediatr. Pulmonol. 43 , 1124–1129 (2008).
Festini, F., Taccetti, G., Cioni, M. L., Repetto, T. & De Martino, M. High incidence of cystic fibrosis in children born in Italy to Albanian immigrants. Thorax 58 , 93 (2003).
Dupuis, A., Hamilton, D., Cole, D. E. & Corey, M. Cystic fibrosis birth rates in Canada: a decreasing trend since the onset of genetic testing. J. Pediatr. 147 , 312–315 (2005).
Kulich, M., Rosenfeld, M., Goss, C. H. & Wilmott, R. Improved survival among young patients with cystic fibrosis. J. Pediatr. 142 , 631–636 (2003).
MacKenzie, T. et al . Longevity of patients with cystic fibrosis in 2000 to 2010 and beyond: survival analysis of the cystic fibrosis foundation patient registry. Ann. Intern. Med. 161 , 233–241 (2014).
Cystic Fibrosis Foundation Cystic Fibrosis Foundation Patient Registry 2012 annual data report. (Cystic Fibrosis Foundation, 2013).
Dodge, J. A., Lewis, P. A., Stanton, M. & Wilsher, J. Cystic fibrosis mortality and survival in the UK: 1947–2003. Eur. Respir. J. 29 , 522–526 (2007). References 15 and 17 show the improvements in survival in the US and the UK cystic fibrosis populations, respectively, over the past decade.
Cystic Fibrosis Canada. Canadian cystic fibrosis patient data registry report 2010. (Cystic Fibrosis Canada, 2010).
Kerem, E. et al . Factors associated with FEV1 decline in cystic fibrosis: analysis of the ECFS patient registry. Eur. Respir. J. 43 , 125–133 (2014).
Rodman, D. M. et al . Late diagnosis defines a unique population of long-term survivors of cystic fibrosis. Am. J. Respir. Crit. Care Med. 171 , 621–626 (2005).
Nick, J. A. et al . Effects of gender and age at diagnosis on disease progression in long-term survivors of cystic fibrosis. Am. J. Respir. Crit. Care Med. 182 , 614–626 (2010).
Simmonds, N. J. et al . Cystic fibrosis and survival to 40 years: a study of cystic fibrosis transmembrane conductance regulator function. Eur. Respir. J. 37 , 1076–1082 (2011).
George, P. M. et al . Improved survival at low lung function in cystic fibrosis: cohort study from 1990 to 2007. BMJ 342 , d1008 (2011).
Accurso, F. J. et al . Effect of VX-770 in persons with cystic fibrosis and the G551D-CFTR mutation. N. Engl. J. Med. 363 , 1991–2003 (2010).
Ramsey, B. W. et al . A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N. Engl. J. Med. 365 , 1663–1672 (2011). This article shows the efficacy of CFTR modulation by the potentiator ivacaftor in patients with the G551D mutation.
Kerem, B. et al . Identification of the cystic fibrosis gene: genetic analysis. Science 245 , 1073–1080 (1989).
Higgins, C. F. ABC transporters: from microorganisms to man. Annu. Rev. Cell Biol. 8 , 67–113 (1992).
Klein, I., Sarkadi, B. & Varadi, A. An inventory of the human ABC proteins. Biochim. Biophys. Acta 1461 , 237–262 (1999).
Collins, F. S. Cystic fibrosis: molecular biology and therapeutic implications. Science 256 , 774–779 (1992).
Serohijos, A. W. et al . Phenylalanine-508 mediates a cytoplasmic-membrane domain contact in the CFTR 3D structure crucial to assembly and channel function. Proc. Natl Acad. Sci. USA 105 , 3256–3261 (2008).
Drumm, M. L. et al . Genetic modifiers of lung disease in cystic fibrosis. N. Engl. J. Med. 353 , 1443–1453 (2005).
Chillon, M. et al . Mutations in the cystic fibrosis gene in patients with congenital absence of the vas deferens. N. Engl. J. Med. 332 , 1475–1480 (1995).
Sharer, N. et al . Mutations of the cystic fibrosis gene in patients with chronic pancreatitis. N. Engl. J. Med. 339 , 645–652 (1998).
Cohn, J. A. et al . Relation between mutations of the cystic fibrosis gene and idiopathic pancreatitis. N. Engl. J. Med. 339 , 653–658 (1998).
Sosnay, P. R. et al . Defining the disease liability of variants in the cystic fibrosis transmembrane conductance regulator gene. Nat. Genet. 45 , 1160–1167 (2013).
Welsh, M. J. & Smith, A. E. Molecular mechanisms of CFTR chloride channel dysfunction in cystic fibrosis. Cell 73 , 1251–1254 (1993).
Okiyoneda, T. et al . Peripheral protein quality control removes unfolded CFTR from the plasma membrane. Science 329 , 805–810 (2010).
Mendoza, J. L. et al . Requirements for efficient correction of DeltaF508 CFTR revealed by analyses of evolved sequences. Cell 148 , 164–174 (2012).
Rabeh, W. M. et al . Correction of both NBD1 energetics and domain interface is required to restore ΔF508 CFTR folding and function. Cell 148 , 150–163 (2012). References 38 and 39 delineate the importance of two distinct molecular defects conferred by the F508del. CFTR mutation, including their effect of correcting CFTR misfolding to restore CFTR expression. These papers show that to fully restore mutated CFTR to the cell surface, both NBD1 stability and interdomain assembly must be addressed.
Green, D. M. et al . Heritability of respiratory infection with Pseudomonas aeruginosa in cystic fibrosis. J. Pediatr. 161 , 290–295 e1 (2012).
Garred, P. et al . Association of mannose-binding lectin gene heterogeneity with severity of lung disease and survival in cystic fibrosis. J. Clin. Invest. 104 , 431–437 (1999).
Sun, L. et al . Multiple apical plasma membrane constituents are associated with susceptibility to meconium ileus in individuals with cystic fibrosis. Nat. Genet. 44 , 562–569 (2012).
Blackman, S. M. et al . Genetic modifiers of cystic fibrosis-related diabetes. Diabetes 62 , 3627–3635 (2013).
Dorfman, R. et al . Modifier gene study of meconium ileus in cystic fibrosis: statistical considerations and gene mapping results. Hum. Genet. 126 , 763–778 (2009).
Romi, H. et al . Meconium ileus caused by mutations in GUCY2C , encoding the CFTR-activating guanylate cyclase 2C. Am. J. Hum. Genet. 90 , 893–899 (2012).
Li, W. et al . Unraveling the complex genetic model for cystic fibrosis: pleiotropic effects of modifier genes on early cystic fibrosis-related morbidities. Hum. Genet. 133 , 151–161 (2014).
Wright, F. A. et al . Genome-wide association and linkage identify modifier loci of lung disease severity in cystic fibrosis at 11p13 and 20q13.2. Nat. Genet. 43 , 539–546 (2011).
Collaco, J. M., Blackman, S. M., McGready, J., Naughton, K. M. & Cutting, G. R. Quantification of the relative contribution of environmental and genetic factors to variation in cystic fibrosis lung function. J. Pediatr. 157 , 802–807 e1–3 (2010).
Vanscoy, L. L. et al . Heritability of lung disease severity in cystic fibrosis. Am. J. Respir. Crit. Care Med. 175 , 1036–1043 (2007).
Kolbe, E. W. et al . CLCA4 variants determine the manifestation of the cystic fibrosis basic defect in the intestine. Eur. J. Hum. Genet. 21 , 691–694 (2013).
Bremer, L. A. et al . Interaction between a novel TGFB1 haplotype and CFTR genotype is associated with improved lung function in cystic fibrosis. Hum. Mol. Genet. 17 , 2228–2237 (2008).
Yarden, J. et al . Association of tumour necrosis factor alpha variants with the CF pulmonary phenotype. Thorax 60 , 320–325 (2005).
Goss, C. H., Newsom, S. A., Schildcrout, J. S., Sheppard, L. & Kaufman, J. D. Effect of ambient air pollution on pulmonary exacerbations and lung function in cystic fibrosis. Am. J. Respir. Crit. Care Med. 169 , 816–821 (2004).
Collaco, J. M. et al . Interactions between secondhand smoke and genes that affect cystic fibrosis lung disease. JAMA 299 , 417–424 (2008).
Martin, B. et al . Comparison of the US and Australian cystic fibrosis registries: the impact of newborn screening. Pediatrics 129 , e348–e355 (2012).
Boyle, M. P., Sabadosa, K. A., Quinton, H. B., Marshall, B. C. & Schechter, M. S. Key findings of the US Cystic Fibrosis Foundation's clinical practice benchmarking project. BMJ Qual. Saf. 23 (Suppl. 1), i15–i22 (2014). This article reports the key outcomes of the quality improvement programme of cystic fibrosis care in the United States and shows the assessment of patterns of practice in the top-performing (20%) treatment centres, and provides insight into the features of practice that could lead to optimization of patient outcomes.
Ooi, C. Y. & Durie, P. R. Cystic fibrosis transmembrane conductance regulator (CFTR) gene mutations in pancreatitis. J. Cyst. Fibros. 11 , 355–362 (2012).
Ratjen, F. & Doring, G. Cystic fibrosis. Lancet 361 , 681–689 (2003).
Rowe, S. M., Miller, S. & Sorscher, E. J. Cystic fibrosis. N. Engl. J. Med. 352 , 1992–2001 (2005).
Boucher, R. C. Airway surface dehydration in cystic fibrosis: pathogenesis and therapy. Annu. Rev. Med. 58 , 157–170 (2007).
Adebamiro, A., Cheng, Y., Rao, U. S., Danahay, H. & Bridges, R. J. A segment of γ-ENaC mediates elastase activation of Na + transport. J. Gen. Physiol. 130 , 611–629 (2007).
Caldwell, R. A., Boucher, R. C. & Stutts, M. J. Neutrophil elastase activates near-silent epithelial Na + channels and increases airway epithelial Na + transport. Am. J. Physiol. Lung Cell. Mol. Physiol. 288 , L813–819 (2005).
Passero, C. J. et al . TMPRSS4-dependent activation of the epithelial sodium channel requires cleavage of the γ-subunit distal to the furin cleavage site. Am. J. Physiol. Renal Physiol. 302 , F1–8 (2012).
Matsui, H. et al . Evidence for periciliary liquid layer depletion, not abnormal ion composition, in the pathogenesis of cystic fibrosis airways disease. Cell 95 , 1005–1015 (1998).
Button, B. et al . A periciliary brush promotes the lung health by separating the mucus layer from airway epithelia. Science 337 , 937–941 (2012).
Chen, J. H. et al . Loss of anion transport without increased sodium absorption characterizes newborn porcine cystic fibrosis airway epithelia. Cell 143 , 911–923 (2010).
Tuggle, K. L. et al . Characterization of defects in ion transport and tissue development in cystic fibrosis transmembrane conductance regulator (CFTR)-knockout rats. PLoS ONE 9 , e91253 (2014).
Itani, O. A. et al . Human cystic fibrosis airway epithelia have reduced Cl − conductance but not increased Na + conductance. Proc. Natl Acad. Sci. USA 108 , 10260–10265 (2011).
Birket, S. E. et al . A functional anatomic defect of the cystic fibrosis airway. Am. J. Respir. Crit. Care Med. 190 , 421–432 (2014).
Hoegger, M. J. et al . Cystic fibrosis. Impaired mucus detachment disrupts mucociliary transport in a piglet model of cystic fibrosis. Science 345 , 818–822 (2014).
Corcoran, T. E. et al . Absorptive clearance of DTPA as an aerosol-based biomarker in the cystic fibrosis airway. Eur. Respir. J. 35 , 781–786 (2010).
Schutte, A. et al . Microbial-induced meprin beta cleavage in MUC2 mucin and a functional CFTR channel are required to release anchored small intestinal mucus. Proc. Natl Acad. Sci. USA 111 , 12396–12401 (2014).
Henderson, A. G. et al . Cystic fibrosis airway secretions exhibit mucin hyperconcentration and increased osmotic pressure. J. Clin. Invest. 124 , 3047–3060 (2014).
Quinton, P. M. Cystic fibrosis: impaired bicarbonate secretion and mucoviscidosis. Lancet 372 , 415–417 (2008).
Garcia, M. A., Yang, N. & Quinton, P. M. Normal mouse intestinal mucus release requires cystic fibrosis transmembrane regulator-dependent bicarbonate secretion. J. Clin. Invest. 119 , 2613–2622 (2009).
Gustafsson, J. K. et al . Bicarbonate and functional CFTR channel are required for proper mucin secretion and link cystic fibrosis with its mucus phenotype. J. Exp. Med. 209 , 1263–1272 (2012). References 70, 71 and 76 demonstrate that a defect in mucociliary clearance emanates from abnormalities of mucus in cystic fibrosis, including its adhesive properties, shown in mouse models in which the intestines are bicarbonate-dependent, porcine airway gland ducts submerged in saline and the human airway surface, even in the setting of adequate periciliary layer depth.
Keiser, N. W. et al . Defective innate immunity and hyper-inflammation in newborn CFTR-knockout ferret lungs. Am. J. Respir. Cell. Mol. Biol. 15 Oct 2014 [epub ahead of print].
Rogers, C. S. et al . Disruption of the CFTR gene produces a model of cystic fibrosis in newborn pigs. Science 321 , 1837–1841 (2008).
Kunzelmann, K. & Mehta, A. CFTR: a hub for kinases and crosstalk of cAMP and Ca 2+ . FEBS J. 280 , 4417–4429 (2013).
Garnett, J. P. et al . Novel role for pendrin in orchestrating bicarbonate secretion in cystic fibrosis transmembrane conductance regulator (CFTR)-expressing airway serous cells. J. Biol. Chem. 286 , 41069–41082 (2011).
Pezzulo, A. A. et al . Reduced airway surface pH impairs bacterial killing in the porcine cystic fibrosis lung. Nature 487 , 109–113 (2012).
Pohl, K. et al . A neutrophil intrinsic impairment affecting Rab27a and degranulation in cystic fibrosis is corrected by CFTR potentiator therapy. Blood 124 , 999–1009 (2014).
Bruscia, E. M. et al . Macrophages directly contribute to the exaggerated inflammatory response in cystic fibrosis transmembrane conductance regulator −/− mice. Am. J. Respir. Cell. Mol. Biol. 40 , 295–304 (2009).
Wang, X. et al . Hsp90 cochaperone Aha1 downregulation rescues misfolding of CFTR in cystic fibrosis. Cell 127 , 803–815 (2006).
Bedrossian, C., Greenberg, S., Singer, D., Hansen, J. & Rosenberg, H. The lung in cystic fibrosis: a quantitative study including prevalence of pathologic findings among different age groups. Hum. Pathol. 7 , 195–204 (1976).
Livraghi-Butrico, A. et al . Mucus clearance, MyD88-dependent and MyD88-independent immunity modulate lung susceptibility to spontaneous bacterial infection and inflammation. Mucosal Immunol. 5 , 397–408 (2012).
Hybiske, K. et al . Effects of cystic fibrosis transmembrane conductance regulator and Delta F508CFTR on inflammatory response, ER stress, and Ca 2+ of airway epithelia. Am. J. Physiol. Lung Cell. Mol. Physiol. 293 , L1250–L1260 (2007).
Mott, L. S. et al . Progression of early structural lung disease in young children with cystic fibrosis assessed using CT. Thorax 67 , 509–516 (2011).
Sly, P. D. et al . Risk factors for bronchiectasis in children with cystic fibrosis. N. Engl. J. Med. 368 , 1963–1970 (2013). This article highlights that neutrophil elastase activity in bronchoalveolar lavage fluid in early life of children with cystic fibrosis undergoing bronchoscopy is associated with early bronchiectasis. This finding shows the importance of early detection of lung infection and inflammation, and the importance of its treatment.
Gehrig, S. et al . Lack of neutrophil elastase reduces inflammation, mucus hypersecretion, and emphysema, but not mucus obstruction, in mice with cystic fibrosis-like lung disease. Am. J. Respir. Crit. Care Med. 189 , 1082–1092 (2014).
Vandivier, R. W. et al . Elastase-mediated phosphatidylserine receptor cleavage impairs apoptotic cell clearance in cystic fibrosis and bronchiectasis. J. Clin. Invest. 109 , 661–670 (2002).
Berger, M., Sorensen, R. U., Tosi, M. F., Dearborn, D. G. & Doring, G. Complement receptor expression on neutrophils at an inflammatory site, the Pseudomonas -infected lung in cystic fibrosis. J. Clin. Invest. 84 , 1302–1313 (1989).
Day, B. J., van Heeckeren, A. M., Min, E. & Velsor, L. W. Role for cystic fibrosis transmembrane conductance regulator protein in a glutathione response to bronchopulmonary pseudomonas infection. Infect. Immun. 72 , 2045–2051 (2004).
Roum, J. H., Buhl, R., McElvaney, N. G., Borok, Z. & Crystal, R. G. Systemic deficiency of glutathione in cystic fibrosis. J. Appl. Physiol. 75 , 2419–2424 (1993).
Knowles, M. R. & Boucher, R. C. Mucus clearance as a primary innate defense mechanism for mammalian airways. J. Clin. Invest. 109 , 571–577 (2002).
Emerson, J., Rosenfeld, M., McNamara, S., Ramsey, B. & Gibson, R. L. Pseudomonas aeruginosa and other predictors of mortality and morbidity in young children with cystic fibrosis. Pediatr. Pulmonol. 34 , 91–100 (2002).
Nixon, G. M. et al . Clinical outcome after early Pseudomonas aeruginosa infection in cystic fibrosis. J. Pediatr. 138 , 699–704 (2001).
Gangell, C. et al . Inflammatory responses to individual microorganisms in the lungs of children with cystic fibrosis. Clin. Infect. Dis. 53 , 425–432 (2011).
van Ewijk, B. E. et al . Prevalence and impact of respiratory viral infections in young children with cystic fibrosis: prospective cohort study. Pediatrics 122 , 1171–1176 (2008).
Wat, D. et al . The role of respiratory viruses in cystic fibrosis. J. Cyst. Fibros. 7 , 320–328 (2008).
Wark, P. A. et al . Viral infections trigger exacerbations of cystic fibrosis in adults and children. Eur. Respir. J. 40 , 510–512 (2012).
Amin, R., Dupuis, A., Aaron, S. D. & Ratjen, F. The effect of chronic infection with Aspergillus fumigatus on lung function and hospitalization in patients with cystic fibrosis. Chest 137 , 171–176 (2010).
Dasenbrook, E. C. et al . Association between respiratory tract methicillin-resistant Staphylococcus aureus and survival in cystic fibrosis. JAMA 303 , 2386–2392 (2010).
Govan, J. R., Brown, A. R. & Jones, A. M. Evolving epidemiology of Pseudomonas aeruginosa and the Burkholderia cepacia complex in cystic fibrosis lung infection. Future Microbiol. 2 , 153–164 (2007).
Ledson, M. J., Gallagher, M. J., Jackson, M., Hart, C. A. & Walshaw, M. J. Outcome of Burkholderia cepacia colonisation in an adult cystic fibrosis centre. Thorax 57 , 142–145 (2002).
Aaron, S. D. et al . Infection with transmissible strains of Pseudomonas aeruginosa and clinical outcomes in adults with cystic fibrosis. JAMA 304 , 2145–2153 (2010).
Armstrong, D. S. et al . Detection of a widespread clone of Pseudomonas aeruginosa in a pediatric cystic fibrosis clinic. Am. J. Respir. Crit. Care Med. 166 , 983–987 (2002).
McCallum, S. J. et al . Superinfection with a transmissible strain of Pseudomonas aeruginosa in adults with cystic fibrosis chronically colonised by P. aeruginosa . Lancet 358 , 558–560 (2001).
Cramer, N. et al . Molecular epidemiology of chronic Pseudomonas aeruginosa airway infections in cystic fibrosis. PLoS ONE 7 , e50731 (2012).
Kidd, T. J. et al . Pseudomonas aeruginosa exhibits frequent recombination, but only a limited association between genotype and ecological setting. PLoS ONE 7 , e44199 (2012).
Wiehlmann, L. et al . Effective prevention of Pseudomonas aeruginosa cross-infection at a cystic fibrosis centre — results of a 10-year prospective study. Int. J. Med. Microbiol. 302 , 69–77 (2012).
Griffiths, A. L. et al . Effects of segregation on an epidemic Pseudomonas aeruginosa strain in a cystic fibrosis clinic. Am. J. Respir. Crit. Care Med. 171 , 1020–1025 (2005).
Adjemian, J., Olivier, K. N. & Prevots, D. R. Nontuberculous mycobacteria among patients with cystic fibrosis in the United States: screening practices and environmental risk. Am. J. Respir. Crit. Care Med. 190 , 581–586 (2014).
Qvist, T. et al . Epidemiology of nontuberculous mycobacteria among patients with cystic fibrosis in Scandinavia. J. Cyst. Fibros. 14 , 46–52 (2014).
Bryant, J. M. et al . Whole-genome sequencing to identify transmission of Mycobacterium abscessus between patients with cystic fibrosis: a retrospective cohort study. Lancet 381 , 1551–1560 (2013).
Rogers, G. B. et al . Bacterial diversity in cases of lung infection in cystic fibrosis patients: 16S ribosomal DNA (rDNA) length heterogeneity PCR and 16S rDNA terminal restriction fragment length polymorphism profiling. J. Clin. Microbiol. 41 , 3548–3558 (2003).
Tunney, M. M. et al . Detection of anaerobic bacteria in high numbers in sputum from patients with cystic fibrosis. Am. J. Respir. Crit. Care Med. 177 , 995–1001 (2008).
LiPuma, J. J. Expanding our understanding of respiratory microbiota in cystic fibrosis. Ann. Am. Thorac. Soc. 11 , 1084–1085 (2014).
Worlitzsch, D. et al . Effects of reduced mucus oxygen concentration in airway Pseudomonas infections of cystic fibrosis patients. J. Clin. Invest. 109 , 317–325 (2002).
Bragonzi, A. et al . Pseudomonas aeruginosa microevolution during cystic fibrosis lung infection establishes clones with adapted virulence. Am. J. Respir. Crit. Care Med. 180 , 138–145 (2009).
Marvig, R. L., Sommer, L. M., Molin, S. & Johansen, H. K. Convergent evolution and adaptation of Pseudomonas aeruginosa within patients with cystic fibrosis. Nat. Genet. 47 , 57–64 (2015).
Young, R. L. et al . Neutrophil extracellular trap (NET)-mediated killing of Pseudomonas aeruginosa : evidence of acquired resistance within the CF airway, independent of CFTR. PLoS ONE 6 , e23637 (2011).
Regamey, N. et al . Distinct patterns of inflammation in the airway lumen and bronchial mucosa of children with cystic fibrosis. Thorax 67 , 164–170 (2012).
Tan, H. L. et al . The Th17 pathway in cystic fibrosis lung disease. Am. J. Respir. Crit. Care Med. 184 , 252–258 (2011).
Hector, A. et al . Regulatory T cell impairment in cystic fibrosis patients with chronic Pseudomonas infection. Am. J. Respir. Crit. Care Med. 191 , 914–923 (2015).
Ooi, C. Y. et al . Does extensive genotyping and nasal potential difference testing clarify the diagnosis of cystic fibrosis among patients with single-organ manifestations of cystic fibrosis? Thorax 69 , 254–260 (2014). This manuscript looks at the use of ancillary testing in the diagnosis of cystic fibrosis. Despite increasing sophistication of ion transport measurements and genetic testing, the diagnosis frequently remains in doubt, underscoring the importance of clinical acumen in these indeterminate cases.
Ooi, C. Y. et al . Comparing the American and European diagnostic guidelines for cystic fibrosis: same disease, different language? Thorax 67 , 618–624 (2012). The article discusses the nomenclature of and the approach to infants with an equivocal newborn screening test. This is a very difficult area of uncertainty for paediatricians and families, and this consensus statement, whether the nomenclature is thought to be correct or not, offers important guidance.
Stewart, B., Zabner, J., Shuber, A. P., Welsh, M. J. & McCray, P. B. Jr. Normal sweat chloride values do not exclude the diagnosis of cystic fibrosis. Am. J. Respir. Crit. Care Med. 151 , 899–903 (1995).
Knowles, M., Gatzy, J. & Boucher, R. Increased bioelectric potential difference across respiratory epithelia in cystic fibrosis. N. Engl. J. Med. 305 , 1489–1495 (1981).
Ooi, C. Y. et al . Does integration of various ion channel measurements improve diagnostic performance in cystic fibrosis? Ann. Am. Thorac Soc. 11 , 562–570 (2014).
Vermeulen, F., Proesmans, M., Boon, M. & De Boeck, K. Improved repeatability of nasal potential difference with a larger surface catheter. J. Cyst. Fibros. [online] , (2014).
Veeze, H. J., Sinaasappel, M., Bijman, J., Bouquet, J. & de Jonge, H. R. Ion transport abnormalities in rectal suction biopsies from children with cystic fibrosis. Gastroenterology 101 , 398–403 (1991).
Cohen-Cymberknoh, M. et al . Evaluation of the intestinal current measurement method as a diagnostic test for cystic fibrosis. Pediatr. Pulmonol. 48 , 229–235 (2013).
Dekkers, J. F. et al . A functional CFTR assay using primary cystic fibrosis intestinal organoids. Nat. Med. 19 , 939–945 (2013).
Quinton, P. et al . β-adrenergic sweat secretion as a diagnostic test for cystic fibrosis. Am. J. Respir. Crit. Care Med. 186 , 732–739 (2012).
Lebecque, P. et al . Mutations of the cystic fibrosis gene and intermediate sweat chloride levels in children. Am. J. Respir. Crit. Care Med. 165 , 757–761 (2002).
Bush, A. & Wallis, C. Time to think again: cystic fibrosis is not an ‘all or none’ disease. Pediatr. Pulmonol. 30 , 139–144 (2000).
Sims, E. J. et al . Economic implications of newborn screening for cystic fibrosis: a cost of illness retrospective cohort study. Lancet 369 , 1187–1195 (2007).
Farrell, P. M. et al . Nutritional benefits of neonatal screening for cystic fibrosis. Wisconsin Cyst. Fibrosis Neonatal Screen. Study Group. N. Engl. J. Med. 337 , 963–969 (1997). This article is a major report on the only randomized controlled trial of screening. Screening led to improved nutritional outcomes but no respiratory improvements. This finding probably reflects suboptimal care in one clinic, making the important point that if cystic fibrosis newborn screening is instituted, the best standards of care for the screen-positive babies must be available.
Article CAS Google Scholar
Wilcken, B. & Gaskin, K. More evidence to favour newborn screening for cystic fibrosis. Lancet 369 , 1146–1147 (2007).
Balfour-Lynn, I. M. Newborn screening for cystic fibrosis: evidence for benefit. Arch. Dis. Child 93 , 7–10 (2008).
Wagener, J. S., Zemanick, E. T. & Sontag, M. K. Newborn screening for cystic fibrosis. Curr. Opin. Pediatr. 24 , 329–335 (2012).
Ranganathan, S. C. et al . Airway function in infants newly diagnosed with cystic fibrosis. Lancet 358 , 1964–1965 (2001).
Ranganathan, S. C. et al . The evolution of airway function in early childhood following clinical diagnosis of cystic fibrosis. Am. J. Respir. Crit. Care Med. 169 , 928–933 (2004).
Southern, K. W. et al . A survey of newborn screening for cystic fibrosis in Europe. J. Cyst. Fibros. 6 , 57–65 (2007).
Vernooij- van Langen, A. M. et al . Novel strategies in newborn screening for cystic fibrosis: a prospective controlled study. Thorax 67 , 289–295 (2012).
Article Google Scholar
Southern, K. W. Determining the optimal newborn screening protocol for cystic fibrosis. Thorax 67 , 281–282 (2012).
Roussey, M. et al . Neonatal screening of cystic fibrosis: diagnostic problems with CFTR mild mutations. J. Inherit Metab. Dis. 30 , 613 (2007).
Scotet, V. et al . Immunoreactive trypsin/DNA newborn screening for cystic fibrosis: should the R117H variant be included in CFTR mutation panels? Pediatrics 118 , e1523–e1529 (2006).
O'Sullivan, B. P., Zwerdling, R. G., Dorkin, H. L., Comeau, A. M. & Parad, R. Early pulmonary manifestation of cystic fibrosis in children with the ΔF508/R117H-7T genotype. Pediatrics 118 , 1260–1265 (2006).
Peckham, D., Conway, S. P., Morton, A., Jones, A. & Webb, K. Delayed diagnosis of cystic fibrosis associated with R117H on a background of 7T polythymidine tract at intron 8. J. Cyst. Fibros. 5 , 63–65 (2006).
de Nooijer, R. A. et al . Assessment of CFTR function in homozygous R117H-7T subjects. J. Cyst. Fibros. 10 , 326–332 (2011).
Cystic Fibrosis, F. et al . Cystic Fibrosis Foundation practice guidelines for the management of infants with cystic fibrosis transmembrane conductance regulator-related metabolic syndrome during the first two years of life and beyond. J. Pediatr. 155 , S106–S116 (2009).
Mayell, S. J. et al . A European consensus for the evaluation and management of infants with an equivocal diagnosis following newborn screening for cystic fibrosis. J. Cyst. Fibros. 8 , 71–78 (2009).
Ren, C. L., Desai, H., Platt, M. & Dixon, M. Clinical outcomes in infants with cystic fibrosis transmembrane conductance regulator (CFTR) related metabolic syndrome. Pediatr. Pulmonol. 46 , 1079–1084 (2011).
Wielputz, M. O. et al . Magnetic resonance imaging detects changes in structure and perfusion, and response to therapy in early cystic fibrosis lung disease. Am. J. Respir. Crit. Care Med. 189 , 956–965 (2014).
Aurora, P. et al . Multiple breath inert gas washout as a measure of ventilation distribution in children with cystic fibrosis. Thorax 59 , 1068–1073 (2004).
Aurora, P. et al . Multiple-breath washout as a marker of lung disease in preschool children with cystic fibrosis. Am. J. Respir. Crit. Care Med. 171 , 249–256 (2005).
Kraemer, R., Blum, A., Schibler, A., Ammann, R. A. & Gallati, S. Ventilation inhomogeneities in relation to standard lung function in patients with cystic fibrosis. Am. J. Respir. Crit. Care Med. 171 , 371–378 (2005).
Lum, S. et al . Early detection of cystic fibrosis lung disease: multiple-breath washout versus raised volume tests. Thorax 62 , 341–347 (2007).
Hoo, A. F. et al . Lung function is abnormal in 3-month-old infants with cystic fibrosis diagnosed by newborn screening. Thorax 67 , 874–881 (2012).
Sly, P. D. et al . Lung disease at diagnosis in infants with cystic fibrosis detected by newborn screening. Am. J. Respir. Crit. Care Med. 180 , 146–152 (2009).
Pillarisetti, N. et al . Infection, inflammation, and lung function decline in infants with cystic fibrosis. Am. J. Respir. Crit. Care Med. 184 , 75–81 (2011).
Nguyen, T. T. et al . Evolution of lung function during the first year of life in newborn screened cystic fibrosis infants. Thorax 69 , 910–917 (2013).
Brennan, L. C. et al . S7 Evolution of lung function during the first two years of life in infants with cystic fibrosis diagnosed by newborn screening. Thorax 68 , A6–A7 (2013).
Amin, R. et al . The effect of dornase alfa on ventilation inhomogeneity in patients with cystic fibrosis. Eur. Respir. J. 37 , 806–812 (2011).
Amin, R. et al . Hypertonic saline improves the LCI in paediatric CF patients with normal lung function. Thorax 65 , 379–383 (2010).
Vermeulen, F., Proesmans, M., Boon, M., Havermans, T. & De Boeck, K. Lung clearance index predicts pulmonary exacerbations in young patients with cystic fibrosis. Thorax 69 , 39–45 (2014).
Maisonneuve, P., Marshall, B. C., Knapp, E. A. & Lowenfels, A. B. Cancer risk in cystic fibrosis: a 20-year nationwide study from the United States. J. Natl Cancer Inst. 105 , 122–129 (2013).
Thia, L. P. et al . Is chest CT useful in newborn screened infants with cystic fibrosis at 1 year of age? Thorax 69 , 320–327 (2014).
Gustafsson, P. M., De Jong, P. A., Tiddens, H. A. & Lindblad, A. Multiple-breath inert gas washout and spirometry versus structural lung disease in cystic fibrosis. Thorax 63 , 129–134 (2008).
Owens, C. M. et al . Lung Clearance Index and HRCT are complementary markers of lung abnormalities in young children with CF. Thorax 66 , 481–488 (2011).
Stafler, P., Davies, J. C., Balfour-Lynn, I. M., Rosenthal, M. & Bush, A. Bronchoscopy in cystic fibrosis infants diagnosed by newborn screening. Pediatr. Pulmonol. 46 , 696–700 (2011).
Wainwright, C. E. et al . Effect of bronchoalveolar lavage-directed therapy on Pseudomonas aeruginosa infection and structural lung injury in children with cystic fibrosis: a randomized trial. JAMA 306 , 163–171 (2011).
CAS PubMed Google Scholar
King, V. V. Upper respiratory disease, sinusitis, and polyposis. Clin. Rev. Allergy 9 , 143–157 (1991).
Henriksson, G. et al . Nasal polyps in cystic fibrosis: clinical endoscopic study with nasal lavage fluid analysis. Chest 121 , 40–47 (2002).
Hansen, S. K. et al . Evolution and diversification of Pseudomonas aeruginosa in the paranasal sinuses of cystic fibrosis children have implications for chronic lung infection. ISME J. 6 , 31–45 (2012).
Rubinstein, S., Moss, R. & Lewiston, N. Constipation and meconium ileus equivalent in patients with cystic fibrosis. Pediatrics 78 , 473–479 (1986).
Blondeau, K. et al . Gastro-oesophageal reflux and aspiration of gastric contents in adult patients with cystic fibrosis. Gut 57 , 1049–1055 (2008).
Ledder, O., Haller, W., Couper, R. T., Lewindon, P. & Oliver, M. Cystic fibrosis: an update for clinicians. Part 2: hepatobiliary and pancreatic manifestations. J. Gastroenterol. Hepatol. 29 , 1954–1962 (2014).
Borowitz, D. et al . Gastrointestinal outcomes and confounders in cystic fibrosis. J. Pediatr. Gastroenterol. Nutr. 41 , 273–285 (2005).
Lewindon, P. J., Robb, T. A., Moore, D. J., Davidson, G. P. & Martin, A. J. Bowel dysfunction in cystic fibrosis: importance of breath testing. J. Paediatr. Child Health 34 , 79–82 (1998).
Konstan, M. W. et al . Growth and nutritional indexes in early life predict pulmonary function in cystic fibrosis. J. Pediatr. 142 , 624–630 (2003).
Sharma, R. et al . Wasting as an independent predictor of mortality in patients with cystic fibrosis. Thorax 56 , 746–750 (2001).
Corbett, K. et al . Cystic fibrosis-associated liver disease: a population-based study. J. Pediatr. 145 , 327–332 (2004).
Rowland, M. et al . Outcome in cystic fibrosis liver disease. Am. J. Gastroenterol. 106 , 104–109 (2011).
Moran, A. et al . Clinical care guidelines for cystic fibrosis-related diabetes: a position statement of the American Diabetes Association and a clinical practice guideline of the Cystic Fibrosis Foundation, endorsed by the Pediatric Endocrine Society. Diabetes Care 33 , 2697–2708 (2010).
Lewis, C. et al . Diabetes-related mortality in adults with cystic fibrosis. Role of genotype and sex. Am. J. Respir. Crit. Care Med. 191 , 194–200 (2015).
Neinstein, L. S., Stewart, D., Wang, C. I. & Johnson, I. Menstrual dysfunction in cystic fibrosis. J. Adolesc. Health Care 4 , 153–157 (1983).
Stern, M., Wiedemann, B., Wenzlaff, P. & German Cystic Fibrosis Quality Assessment, G. From registry to quality management: the German Cystic Fibrosis Quality Assessment project 1995–2006. Eur. Respir. J. 31 , 29–35 (2008).
Marshall, B. C. & Nelson, E. C. Accelerating implementation of biomedical research advances: critical elements of a successful 10 year Cystic Fibrosis Foundation healthcare delivery improvement initiative. BMJ Qual. Saf. 23 (Suppl. 1), i95–i103 (2014).
McCormick, J. et al . Comparative demographics of the European cystic fibrosis population: a cross-sectional database analysis. Lancet 375 , 1007–1013 (2010).
Colombo, C. & Littlewood, J. The implementation of standards of care in Europe: state of the art. J. Cyst. Fibros. 10 (Suppl. 2), S7–S15 (2011).
Stern, M., Niemann, N., Wiedemann, B., Wenzlaff, P. & German, C. G. Benchmarking improves quality in cystic fibrosis care: a pilot project involving 12 centres. Int. J. Qual. Health Care 23 , 349–356 (2011).
Schechter, M. S. Benchmarking to improve the quality of cystic fibrosis care. Curr. Opin. Pulm. Med. 18 , 596–601 (2012).
Conway, S. et al . European Cystic Fibrosis Society standards of care: framework for the Cystic Fibrosis Centre. J. Cyst. Fibros. 13 , S3–S22 (2014).
European Cystic Fibrosis Society. ECFS Patient Registry annual data report 2010. (European Cystic Fibrosis Society, 2010).
Cystic Fibrosis Trust. UK CF Registry annual report 2011. (Cystic Fibrosis Trust, 2013).
Goss, C. H. et al . Children and young adults with CF in the USA have better lung function compared with the UK. Thorax 70 , 229–236 (2014).
Cystic Fibrosis Australia. 15th Annual Report from the Cystic Fibrosis Data Registry. (Cystic Fibrosis Australia, 2013).
Stern, M. et al . European Cystic Fibrosis Society standards of care: quality management in cystic fibrosis. J. Cyst. Fibros. 13 , S43–S59 (2014).
Quittner, A. L. et al . Pulmonary medication adherence and health-care use in cystic fibrosis. Chest 146 , 142–151 (2014).
Abbott, J., Dodd, M. & Webb, A. K. Health perceptions and treatment adherence in adults with cystic fibrosis. Thorax 51 , 1233–1238 (1996).
Conway, S. P., Pond, M. N., Hamnett, T. & Watson, A. Compliance with treatment in adult patients with cystic fibrosis. Thorax 51 , 29–33 (1996).
Plant, B. J., Goss, C. H., Plant, W. D. & Bell, S. C. Management of comorbidities in older patients with cystic fibrosis. Lancet Respir. Med. 1 , 164–174 (2013).
Grasemann, H. & Ratjen, F. Early lung disease in cystic fibrosis. Lancet Respir. Med. 1 , 148–157 (2013).
Flume, P. A. et al . Cystic fibrosis pulmonary guidelines: treatment of pulmonary exacerbations. Am. J. Respir. Crit. Care Med. 180 , 802–808 (2009).
Smyth, A. R. et al . European Cystic Fibrosis Society Standards of Care: Best Practice guidelines. J. Cyst. Fibros. 13 , S23–S42 (2014).
Okumura, M. J. et al . Improving transition from paediatric to adult cystic fibrosis care: programme implementation and evaluation. BMJ Qual. Saf. 23 (Suppl. 1), i64–i72 (2014).
Lester, M. K. & Flume, P. A. Airway-clearance therapy guidelines and implementation. Respir. Care 54 , 733–753 (2009).
McIlwaine, M. P. et al . Long-term multicentre randomised controlled study of high frequency chest wall oscillation versus positive expiratory pressure mask in cystic fibrosis. Thorax 68 , 746–751 (2013).
Schneiderman, J. E. et al . Longitudinal relationship between physical activity and lung health in patients with cystic fibrosis. Eur. Respir. J. 43 , 817–823 (2014).
Cox, N. S., Alison, J. A. & Holland, A. E. Interventions for promoting physical activity in people with cystic fibrosis. Cochrane Database Syst. Rev. 12 , CD009448 (2013).
Fuchs, H. J. et al . Effect of aerosolized recombinant human DNase on exacerbations of respiratory symptoms and on pulmonary function in patients with cystic fibrosis. N. Engl. J. Med. 331 , 637–642 (1994).
Jones, A. P. & Wallis, C. E. Recombinant human deoxyribonuclease for cystic fibrosis. Cochrane Database Syst. Rev. 3 , CD001127 (2003).
Shah, P. I. et al . Recombinant human DNase I in cystic fibrosis patients with severe pulmonary disease: a short-term, double-blind study followed by six months open-label treatment. Eur. Respir. J. 8 , 954–958 (1995).
Quan, J. M. et al . A two-year randomized, placebo-controlled trial of dornase alfa in young patients with cystic fibrosis with mild lung function abnormalities. J. Pediatr. 139 , 813–820 (2001).
Flume, P. A. et al . Cystic fibrosis pulmonary guidelines: chronic medications for maintenance of lung health. Am. J. Respir. Crit. Care Med. 176 , 957–969 (2007). References 197 and 218 describe current standards of care of implementing a cystic fibrosis centre, and for the management of patients with chronic maintenance therapies, respectively.
Elkins, M. R. et al . A controlled trial of long-term inhaled hypertonic saline in patients with cystic fibrosis. N. Engl. J. Med. 354 , 229–240 (2006).
Rosenfeld, M. et al . Inhaled hypertonic saline in infants and children younger than 6 years with cystic fibrosis: the ISIS randomized controlled trial. JAMA 307 , 2269–2277 (2012).
Subbarao, P. et al . Lung clearance index as an outcome measure for clinical trials in young children with cystic fibrosis. A pilot study using inhaled hypertonic saline. Am. J. Respir. Crit. Care Med. 188 , 456–460 (2013).
Bilton, D. et al . Inhaled dry powder mannitol in cystic fibrosis: an efficacy and safety study. Eur. Respir. J. 38 , 1071–1080 (2011).
Aitken, M. L. et al . Long-term inhaled dry powder mannitol in cystic fibrosis: an international randomized study. Am. J. Respir. Crit. Care Med. 185 , 645–652 (2012).
Ramsey, B. W. et al . Efficacy of aerosolized tobramycin in patients with cystic fibrosis. N. Engl. J. Med. 328 , 1740–1746 (1993).
Ramsey, B. W. et al . Intermittent administration of inhaled tobramycin in patients with cystic fibrosis. N. Engl. J. Med. 340 , 23–30 (1999).
Konstan, M. W. et al . Safety, efficacy and convenience of tobramycin inhalation powder in cystic fibrosis patients: The EAGER trial. J. Cyst. Fibros. 10 , 54–61 (2011).
McCoy, K. S. et al . Inhaled aztreonam lysine for chronic airway Pseudomonas aeruginosa in cystic fibrosis. Am. J. Respir. Crit. Care Med. 178 , 921–928 (2008).
Retsch-Bogart, G. Z. et al . A phase 2 study of aztreonam lysine for inhalation to treat patients with cystic fibrosis and Pseudomonas aeruginosa infection. Pediatr. Pulmonol. 43 , 47–58 (2008).
Doring, G., Flume, P., Heijerman, H., Elborn, J. S. & Consensus Study, G. Treatment of lung infection in patients with cystic fibrosis: current and future strategies. J. Cyst. Fibros. 11 , 461–479 (2012).
Tullis, D. E. et al . Inhaled aztreonam for chronic Burkholderia infection in cystic fibrosis: a placebo-controlled trial. J. Cyst. Fibros. 13 , 296–305 (2014).
Wolter, J. et al . Effect of long term treatment with azithromycin on disease parameters in cystic fibrosis: a randomised trial. Thorax 57 , 212–216 (2002).
Saiman, L. et al . Azithromycin in patients with cystic fibrosis chronically infected with Pseudomonas aeruginosa : a randomized controlled trial. JAMA 290 , 1749–1756 (2003).
Saiman, L. et al . Effect of azithromycin on pulmonary function in patients with cystic fibrosis uninfected with Pseudomonas aeruginosa : a randomized controlled trial. JAMA 303 , 1707–1715 (2010).
Clement, A. et al . Long term effects of azithromycin in patients with cystic fibrosis: A double blind, placebo controlled trial. Thorax 61 , 895–902 (2006).
Konstan, M. W., Byard, P. J., Hoppel, C. L. & Davis, P. B. Effect of high-dose ibuprofen in patients with cystic fibrosis. N. Engl. J. Med. 332 , 848–854 (1995).
Lands, L. C. & Stanojevic, S. Oral non-steroidal anti-inflammatory drug therapy for lung disease in cystic fibrosis. Cochrane Database Syst, Rev. 6 , CD001505 (2013).
Auerbach, H. S., Williams, M., Kirkpatrick, J. A. & Colten, H. R. Alternate-day prednisone reduces morbidity and improves pulmonary function in cystic fibrosis. Lancet 2 , 686–688 (1985).
Eigen, H., Rosenstein, B. J., FitzSimmons, S. & Schidlow, D. V. A multicenter study of alternate-day prednisone therapy in patients with cystic fibrosis. J. Pediatr. 126 , 515–523 (1995).
Balfour-Lynn, I. M. & Welch, K. Inhaled corticosteroids for cystic fibrosis. Cochrane Database Syst. Rev. 11 , CD001915 (2009).
Balfour-Lynn, I. M. et al . Multicenter randomized controlled trial of withdrawal of inhaled corticosteroids in cystic fibrosis. Am. J. Respir. Crit. Care Med. 173 , 1356–1362 (2006).
Konstan, M. W. et al . A randomized double blind, placebo controlled phase 2 trial of BIIL 284 BS (an LTB4 receptor antagonist) for the treatment of lung disease in children and adults with cystic fibrosis. J. Cyst. Fibros. 13 , 148–155 (2014).
Hirche, T. O. et al . Practical guidelines: lung transplantation in patients with cystic fibrosis. Pulm. Med. 2014 , 621342 (2014).
Madden, B. P. et al . Noninvasive ventilation in cystic fibrosis patients with acute or chronic respiratory failure. Eur. Respir. J. 19 , 310–313 (2002).
Gill, D. R. & Hyde, S. C. Delivery of genes into the CF airway. Thorax 69 , 962–964 (2014).
Crystal, R. G. et al . Administration of an adenovirus containing the human CFTR cDNA to the respiratory tract of individuals with cystic fibrosis. Nat. Genet. 8 , 42–51 (1994).
Harvey, B. G., Hackett, N. R., Ely, S. & Crystal, R. G. Host responses and persistence of vector genome following intrabronchial administration of an E1 − E3 − , adenovirus gene transfer vector to normal individuals. Mol. Ther. 3 , 206–215 (2001).
Knowles, M. R. et al . A controlled study of adenoviral-vector-mediated gene transfer in the nasal epithelium of patients with cystic fibrosis. N. Engl. J. Med. 333 , 823–831 (1995).
Moss, R. B. et al . Repeated adeno-associated virus serotype 2 aerosol-mediated cystic fibrosis transmembrane regulator gene transfer to the lungs of patients with cystic fibrosis: a multicenter, double-blind, placebo-controlled trial. Chest 125 , 509–521 (2004).
Moss, R. B. et al . Repeated aerosolized AAV-CFTR for treatment of cystic fibrosis: a randomized placebo-controlled phase 2B trial. Hum. Gene Ther. 18 , 726–732 (2007).
Griesenbach, U. et al . Assessment of F/HN-pseudotyped lentivirus as a clinically relevant vector for lung gene therapy. Am. J. Respir. Crit. Care Med. 186 , 846–856 (2012).
Alton, E. W. et al . A randomised, double-blind, placebo-controlled phase IIB clinical trial of repeated application of gene therapy in patients with cystic fibrosis. Thorax 68 , 1075–1077 (2013).
Bangel-Ruland, N. et al . Cystic fibrosis transmembrane conductance regulator-mRNA delivery: a novel alternative for cystic fibrosis gene therapy. J. Gene Med. 15 , 414–426 (2013).
Wilschanski, M. et al . Gentamicin-induced correction of CFTR function in patients with cystic fibrosis and CFTR stop mutations. N. Engl. J. Med. 349 , 1433–1441 (2003).
Linde, L. et al . Nonsense-mediated mRNA decay affects nonsense transcript levels and governs response of cystic fibrosis patients to gentamicin. J. Clin. Invest. 117 , 683–692 (2007).
Kerem, E. et al . Ataluren for the treatment of nonsense-mutation cystic fibrosis: a randomised, double-blind, placebo-controlled phase 3 trial. Lancet Respir. Med. 2 , 539–547 (2014).
Study of ataluren (PTC124 ® ) in cystic fibrosis [online] , (2014).
Van Goor, F. et al . Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc. Natl Acad. Sci. USA 106 , 18825–18830 (2009).
Rowe, S. M. et al . Clinical mechanism of the cystic fibrosis transmembrane conductance regulator potentiator ivacaftor in G551D-mediated cystic fibrosis. Am. J. Respir. Crit. Care Med. 190 , 175–184 (2014).
De Boeck, K. et al . Efficacy and safety of ivacaftor in patients with cystic fibrosis and a non-G551D gating mutation. J. Cyst. Fibros. 13 , 674–680 (2014).
Sloane, P. A. et al . A pharmacologic approach to acquired cystic fibrosis transmembrane conductance regulator dysfunction in smoking related lung disease. PLoS ONE 7 , e39809 (2012).
Flume, P. A. et al . Ivacaftor in subjects with cystic fibrosis who are homozygous for the F508del-CFTR mutation. Chest 142 , 718–724 (2012).
Boyle, M. P. et al . A CFTR corrector (lumacaftor) and a CFTR potentiator (ivacaftor) for treatment of patients with cystic fibrosis who have a phe508del CFTR mutation: a phase 2 randomised controlled trial 2 , 527–538 (2014).
Van Goor, F. et al . Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proc. Natl Acad. Sci. USA 108 , 18843–18848 (2011).
Veit, G. et al . Some gating potentiators, including VX-770, diminish ΔF508–CFTR functional expression. Sci. Transl. Med. 6 , 246ra97 (2014).
Cholon, D. M. et al . Potentiator ivacaftor abrogates pharmacological correction of ΔF508 CFTR in cystic fibrosis. Sci. Transl. Med. 6 , 246ra96 (2014).
Okiyoneda, T. et al . Mechanism-based corrector combination restores DeltaF508-CFTR folding and function. Nat. Chem. Biol. 9 , 444–454 (2013).
Accurso, F. J. et al . Denufosol tetrasodium in patients with cystic fibrosis and normal to mildly impaired lung function. Am. J. Respir. Crit. Care Med. 183 , 627–634 (2011).
Ratjen, F. et al . Long term effects of denufosol tetrasodium in patients with cystic fibrosis. J. Cyst. Fibros. 11 , 539–549 (2012).
Burrows, E. F., Southern, K. W. & Noone, P. G. Sodium channel blockers for cystic fibrosis. Cochrane Database Syst, Rev. 4 , CD005087 (2014).
Quittner, A. L., Alpern, A. N. & Kimberg, C. I. in Kendig and Chernick's Disorders of the Respiratory Tract in Children . 8th edn (eds Wilmott, R. W., Boat, T. F., Bush, A., Chernick, V., Deterding, R. R. & Ratjen, F. ) 251–260 (Elsevier (Saunders), 2012).
Book Google Scholar
Goss, C. H., Edwards, T. C., Ramsey, B. W., Aitken, M. L. & Patrick, D. L. Patient-reported respiratory symptoms in cystic fibrosis. J. Cyst. Fibros. 8 , 245–252 (2009).
Schmidt, A. M. et al . Exercise and quality of life in patients with cystic fibrosis: A 12-week intervention study. Physiother Theory Pract. 27 , 548–556 (2011).
Schechter, M. S. et al . Long-term effects of pregnancy and motherhood on disease outcomes of women with cystic fibrosis. Ann. Am. Thorac Soc. 10 , 213–219 (2013).
Sawicki, G. S. et al . Longitudinal assessment of health-related quality of life in an observational cohort of patients with cystic fibrosis. Pediatr. Pulmonol. 46 , 36–44 (2011).
Snyder, C. F. et al . Implementing patient-reported outcomes assessment in clinical practice: a review of the options and considerations. Qual. Life Res. 21 , 1305–1314 (2012).
Sawicki, G. S., Sellers, D. E. & Robinson, W. M. High treatment burden in adults with cystic fibrosis: challenges to disease self-management. J. Cyst. Fibros. 8 , 91–96 (2009).
Quittner, A. L. Measurement of quality of life in cystic fibrosis. Curr. Opin. Pulm. Med. 4 , 326–331 (1998).
Goss, C. H. & Quittner, A. L. Patient-reported outcomes in cystic fibrosis. Proc. Am. Thorac. Soc. 4 , 378–386 (2007).
Quittner, A. L. et al . Erratum to: Psychometric evaluation of the Cystic Fibrosis Questionnaire-Revised in a national, US sample. Qual. Life Res. 21 , 1279–1290 (2012).
Donaldson, S. H. et al . Mucus clearance and lung function in cystic fibrosis with hypertonic saline. N. Engl. J. Med. 354 , 241–250 (2006).
Sawicki, G. S. et al . Treatment complexity in cystic fibrosis: trends over time and associations with site-specific outcomes. J. Cyst. Fibros. 12 , 461–467 (2013). References 277 and 281 show the treatment burden and the complexity in patients with cystic fibrosis, respectively.
Quittner, A. L. et al . Impact of socioeconomic status, race, and ethnicity on quality of life in patients with cystic fibrosis in the United States. Chest 137 , 642–650 (2010).
Quittner, A. L. et al . Prevalence of depression and anxiety in patients with cystic fibrosis and parent caregivers: results of The International Depression Epidemiological Study across nine countries. Thorax 69 , 1090–1097 (2014).
Smith, B. A., Modi, A. C., Quittner, A. L. & Wood, B. L. Depressive symptoms in children with cystic fibrosis and parents and its effects on adherence to airway clearance. Pediatr. Pulmonol. 45 , 756–763 (2010).
Riekert, K. A., Bartlett, S. J., Boyle, M. P., Krishnan, J. A. & Rand, C. S. The association between depression, lung function, and health-related quality of life among adults with cystic fibrosis. Chest 132 , 231–237 (2007).
Goldbeck, L. et al . Prevalence of symptoms of anxiety and depression in German patients with cystic fibrosis. Chest 138 , 929–936 (2010).
Snell, C., Fernandes, S., Bujoreanu, I. S. & Garcia, G. Depression, illness severity, and healthcare utilization in cystic fibrosis. Pediatr. Pulmonol. 49 , 1177–1181 (2014).
Wong, A. P. et al . Directed differentiation of human pluripotent stem cells into mature airway epithelia expressing functional CFTR protein. Nat. Biotech. 30 , 876–882 (2012). References 135 and 288 describe model systems to assess response to CFTR modulators in vitro by either using intestinal organoids or skin-derived pluripotent stem cells transformed into airway epithelial cells, respectively.
Download references
Author information
Authors and affiliations.
Division of Respiratory Medicine, Department of Paediatrics, The Hospital for Sick Children and University of Toronto, 555 University Avenue, Toronto, M5G 1X8, Canada
Felix Ratjen
Department of Thoracic Medicine, Queensland Children's Medical Research Institute, Brisbane, Queensland, Australia
Scott C. Bell
Department of Medicine, University of Alabama at Birmingham, Birmingham, Alabama, USA
Steven M. Rowe
Division of Pulmonary and Critical Care Medicine, University of Washington Medical Center, Seattle, Washington, USA
Christopher H. Goss
Department of Psychology, University of Miami, Miami, Florida, USA
Alexandra L. Quittner
Paediatrics Section, Imperial College, Paediatric Respirology, National Heart and Lung Institute, and the Royal Brompton Harefield NHS Foundation Trust, London, UK
Andrew Bush
You can also search for this author in PubMed Google Scholar
Contributions
Introduction (F.R.); Epidemiology (C.H.G.), Mechanisms/pathophysiology (S.M.R. and S.C.B.); Diagnosis, screening and prevention (A.B., F.R. and S.M.R.); Management (S.C.B. and F.R.); Quality of life (A.L.Q.); Outlook (F.R. and A.B.); overview of the Primer (F.R.).
Corresponding author
Correspondence to Felix Ratjen .
Ethics declarations
Competing interests.
S.M.R. has received grants and/or non-financial support from: Cystic Fibrosis Foundation Therapeutics, the US National Institutes of Health (NIH), Vertex Pharmaceuticals, PTC Therapeutics, Novartis, Forest Research Institute, Bayer Healthcare and Galapagos. C.H.G. has received grant funding and/or honoraria from: the NIH (grants P30 DK089507, R01HL103965, R01AI101307 and UM1HL119073), Food and Drug Administration (grant R01FD003704), the Cystic Fibrosis Foundation, Vertex Pharmaceuticals, Transave Inc., L. Hoffmann-La Roche Ltd, Johns Hopkins University, the European Cystic Fibrosis Society, Medscape and Gilead Sciences. He has also participated in Advisory Boards for KaloBios Pharmaceuticals and Transave Inc. A.L.Q. has received grants and/or consulting income from: NIH (grant R01 DC04797), European Union Health Commission (BESTCILIA), National Health and Medical Research Council of Australia, Cystic Fibrosis Foundation Clinical Research Grant, Novartis, Abbott Pharmaceuticals, Vertex Pharmaceuticals and Gilead Sciences. F.R. has received grants and/or consulting fees from: the Canadian Institutes of Health Research, National Heart, Lung, and Blood Institute, the Cystic Fibrosis Foundation, Genentech, Vertex Pharmaceuticals, Novartis, Gilead Sciences, Boehringer Ingelheim and Roche. S.C.B. has received grants, personal fees, speaker's fees and/or non-financial support from the National Health and Medical Research Council of Australia, the Cystic Fibrosis Foundation, the Office of Health and Medical Research, Queensland Health, the Queensland Children's Foundation, Vertex Pharmaceuticals, Novartis and Gilead. He has served on advisory boards for Vertex Pharmaceuticals, Novartis and Rempex and as a site principal investigator in several clinical trials sponsored by Vertex Pharmaceuticals. A.B. is supported by the UK National Institute of Health Research Respiratory Disease Biomedical Research Unit at the Royal Brompton and Harefield National Health Service Foundation Trust and Imperial College London.
PowerPoint slides
Powerpoint slide for fig. 1, powerpoint slide for fig. 2, powerpoint slide for fig. 3, powerpoint slide for fig. 4, powerpoint slide for fig. 5, powerpoint slide for fig. 6, rights and permissions.
Reprints and permissions
About this article
Cite this article.
Ratjen, F., Bell, S., Rowe, S. et al. Cystic fibrosis. Nat Rev Dis Primers 1 , 15010 (2015). https://doi.org/10.1038/nrdp.2015.10
Download citation
Published : 14 May 2015
DOI : https://doi.org/10.1038/nrdp.2015.10
Share this article
Anyone you share the following link with will be able to read this content:
Sorry, a shareable link is not currently available for this article.
Provided by the Springer Nature SharedIt content-sharing initiative
This article is cited by
Solvent effects in anion recognition.
- Sophie C. Patrick
- Paul D. Beer
- Jason J. Davis
Nature Reviews Chemistry (2024)
Dual-color live imaging unveils stepwise organization of multiple basal body arrays by cytoskeletons
- Gen Shiratsuchi
- Satoshi Konishi
- Sachiko Tsukita
EMBO Reports (2024)
Longitudinal microbial and molecular dynamics in the cystic fibrosis lung after Elexacaftor–Tezacaftor–Ivacaftor therapy
- Christian Martin
- Douglas V. Guzior
- Robert A. Quinn
Respiratory Research (2023)
Lung development and regeneration: newly defined cell types and progenitor status
- Xiaogao Meng
- Guizhong Cui
- Guangdun Peng
Cell Regeneration (2023)
Fractional Exhalation Nitric Oxide (FeNO) changes in cystic fibrosis patients induced by compound honey syrup: a pretest–posttest clinical trial
- Hanieh Tahermohammadi
- Shima Derikvandi
BMC Pulmonary Medicine (2023)
Quick links
- Explore articles by subject
- Guide to authors
- Editorial policies
Sign up for the Nature Briefing newsletter — what matters in science, free to your inbox daily.

Cystic fibrosis occurs when the cystic fibrosis transmembrane conductance regulator (CFTR) protein is either not made correctly, or not made at all. By understanding how the protein is made, scientists have been able to develop treatments that target the protein and restore its function.
- The cystic fibrosis transmembrane conductance regulator (CFTR) protein helps to maintain the balance of salt and water on many surfaces in the body, such as the surface of the lung.
- The CFTR protein is a particular type of protein called an ion channel. In the lung, the CFTR ion channel moves chloride ions from inside the cell to outside the cell.
Researchers are still trying to learn more about the structure of the CFTR protein so that they can find new and better ways to help improve the function of the protein in people with CF.
The cystic fibrosis transmembrane conductance regulator (CFTR) protein helps to maintain the balance of salt and water on many surfaces in the body, such as the surface of the lung. When the protein is not working correctly, chloride — a component of salt — becomes trapped in cells. Without the proper movement of chloride, water cannot hydrate the cellular surface. This leads the mucus covering the cells to become thick and sticky, causing many of the symptoms associated with cystic fibrosis.
To understand how mutations in the CFTR gene cause the protein to become dysfunctional, it is important to understand how the protein is normally made, and how it helps to move water and chloride to the cell surface.
What Are Proteins?
Proteins are tiny machines that do specific jobs within a cell. The instructions for building each protein are encoded in DNA. Proteins are assembled from building blocks called amino acids. There are 20 different amino acids. All proteins are made up of chains of these amino acids connected together in different orders, like different words that are written using the same 26 letters of the alphabet. The DNA instructions tell the cell which amino acid to use at each position in the chain to make a specific protein.
The CFTR protein is made up of 1,480 amino acids. Once the CFTR protein chain is made, it is folded into a specific 3-D shape. The CFTR protein is shaped like a tube that goes through the membrane surrounding the cell, like a straw goes through the plastic top on a cup.

What Does the CFTR Protein Do?
The CFTR protein is a particular type of protein called an ion channel. An ion channel moves atoms or molecules that have an electrical charge from inside the cell to outside, or from outside the cell to inside. In the lung, the CFTR ion channel moves chloride ions from inside the cell to outside the cell. To get out of the cell, the chloride ions move through the center of the tube formed by the CFTR protein.
Once the chloride ions are outside the cell, they attract a layer of water. This water layer is important because it allows tiny hairs on the surface of the lung cells, called cilia, to sweep back and forth. This sweeping motion moves mucus up and out of the airways.
How Do Problems With the CFTR Protein Cause CF?
In people with CF, mutations in the CFTR gene can cause the following problems with the CFTR protein:
- It doesn't work well
- It isn't produced in sufficient quantities
- It is not produced at all
When any of these problems occur, the chloride ions are trapped inside the cell, and water is no longer attracted to the space outside the cell. When there is less water outside the cells, the mucus in the airways becomes dehydrated and thickens, causing it to flatten the cilia. The cilia can't sweep properly when thick, sticky mucus weighs them down.
Because the cilia can't move properly, mucus gets stuck in the airways, making it difficult to breathe. In addition, germs caught in the mucus are no longer expelled from the airway, allowing them to multiply and cause infections. Thick mucus in the lungs and frequent airway infections are some of the most common problems people with CF face.
Researchers Are Still Studying the Basic Structure

Because the 3-D shape of CFTR is so complex, it was not until early 2017 that the first high-resolution pictures were developed. These pictures have given researchers important clues about where drugs bind the protein, how they affect its function, and how to develop new CF therapies. In the future, pictures showing the protein in an “open” position, where salt can move through, will be even more helpful to researchers developing new CF therapies.
CF Genetics: The Basics Article | 6 min read
Types of CFTR Mutations Article | 9 min read
Restore CFTR: Exploring Treatments for Rare and Nonsense Mutations Article | 8 min read
Find Out More About Your Mutations Article | 3 min read

( 1-800-344-4823 ) Mon - Thu, 9 am - 5 pm ET Fri, 9 am - 3 pm ET
An official website of the United States government
The .gov means it’s official. Federal government websites often end in .gov or .mil. Before sharing sensitive information, make sure you’re on a federal government site.
The site is secure. The https:// ensures that you are connecting to the official website and that any information you provide is encrypted and transmitted securely.
- Publications
- Account settings
Preview improvements coming to the PMC website in October 2024. Learn More or Try it out now .
- Advanced Search
- Journal List
- v.13(7); 2021 Jul
A Review of Trikafta: Triple Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Modulator Therapy
1 Internal Medicine, University of Debrecen, Debrecen, HUN
Jude ElSaygh
Dalal elsori.
2 Pediatrics, Rhode Island Hospital, Brown University, Rhode Island, USA
Hassan ElSaygh
Abdulsabar sanni.
3 Internal Medicine, Hennepin Healthcare, Minnesota, USA
Cystic fibrosis (CF) is a potentially fatal genetic disease that causes serious lung damage. With time, researchers have a more complete understanding of the molecular-biological defects that underlie CF. This knowledge is leading to alternative approaches regarding the treatment of this condition. Trikafta is the third FDA-approved drug that targets the F508del mutation of the CFTR gene. The drug is a combination of three individual drugs which are elexacaftor (ELX), tezacaftor (TEZ), and ivacaftor (IVA). This trio increases the activity of the cystic fibrosis transmembrane conductance regulator (CFTR) protein and reduces the mortality and morbidity rates in CF patients. The effectiveness of Trikafta, seen in clinical trials, outperforms currently available therapies in terms of lung function, quality of life, sweat chloride reduction, and pulmonary exacerbation reduction. The safety and efficacy of CFTR modulators in children with CF have also been studied. Continued evaluation of patient data is needed to confirm its long-term safety and efficacy. In this study, we will focus on reviewing data from clinical trials regarding the benefits of CFTR modulator therapy. We address the impact of Trikafta on lung function, pulmonary exacerbations, and quality of life. Adverse events of the different CFTR modulators are discussed.
Introduction and background
Cystic fibrosis (CF) is a chronic, progressive, autosomal recessive disease that affects approximately 35,000 people in the United States [ 1 , 2 ]. The primary defect is a mutant CFTR gene causing a decrease or absence of cystic fibrosis transmembrane conductance regulator (CFTR) activity. CFTR protein is an epithelial anion transporter of chloride and bicarbonate. It regulates salt and water balance on the surface of cells and is encoded by the CFTR gene. A defect in the CFTR protein will cause pulmonary, gastrointestinal, pancreatic, and reproductive system diseases [ 3 ].
In CF, the CFTR protein is often expressed in epithelial cells' apical membranes. In CF patients, the secreted fluids are mostly mucus- and protein-rich [ 4 ]. CFTR proteins are expressed in the airway epithelia, para-nasal sinuses, pancreas, gut epithelia, biliary tree epithelia, vas deferens epithelia, and sweat duct epithelia [ 5 ]. However, the airway epithelia carry the highest levels of CFTR expression.
More than 1500 mutations in the CFTR gene have been discovered in the northern European and North American populations. Different mutations are divided into the 6 classes illustrated in Table Table1. 1 . Class II mutations account for around two-thirds of the mutations found in CF patients. Usually, it’s due to the deletion of phenylalanine at the location 508 allele [ 2 , 6 ]. Protein misfolding and retention at the endoplasmic reticulum (ER) by the ER quality control system also affect the CFTR protein flow. Premature degradation occurs after such retention, preventing the protein from reaching the cell surface and severely reducing CFTR function [ 7 ].
| Type of mutation | Type of CFTR mutation | Percent of people with CF who have at least 1 mutations. |
| Normal | CFTR protein is created and moves to the cell surface, allowing the transfer of chloride and water. | |
| Class I | No functional CFTR protein is created | 22 percent |
| Class II | CFTR protein is created but misfolds, keeping it from moving to the cell surface. This is called a trafficking defect. | 88 percent |
| Class III | CFTR protein is created and moves to the cell surface but the channel gate does not open. This is called a defective channel regulation. | 6 percent |
| Class IV | CFTR protein is created and moves to the cell surface but the channel function is faulty. This is called decreased channel conductance. | 6 percent |
| Class V | Normal CFTR protein is created and moves correctly to the cell surface but not enough amount of the protein. This is called reduced synthesis of CFTR. | 5 percent |
| Class VI | CFTR protein is created but it does not work properly at the cell membrane. This is called decreased CFTR stability. | 5 percent |
In the absence of a functioning CFTR, cyclic adenosine monophosphate (cAMP)-dependent chloride and bicarbonate secretion into airway secretions will be impaired. Mucins become tethered to the bronchial apical surfaces and create an easy nidus for bacteria to invade [ 4 ]. Progressive lung failure due to opportunistic pathogen infection and mucosal chronic inflammation is a major source of morbidity and mortality in CF patients. CFTR protein deficiency also causes progressive fat and vitamin malabsorption and the inability to thrive [ 4 ].
As mentioned above, the major cause of morbidity and mortality in CF is respiratory failure [ 8 ]. Most patients ultimately develop progressive lung disease with airway mucus obstruction, bacterial infection, and inflammation despite intensive symptomatic therapies. Symptomatic therapies do not address the disease's molecular cause, often causing the disease to progress and complications to arise. Treatments that target the CFTR molecular defect are needed to halt the chain of events that leads to progressive lung disease [ 7 ]. It is important to note that new evidence surfaced suggesting that the CFTR protein plays an important role in many major respiratory disorders, such as asthma and chronic obstructive pulmonary disease [ 7 ].
A search was performed on databases including PubMed, Google scholar, and Journal of the European Cystic Fibrosis Society using keywords such as CF, ivacaftor (IVA), lumacaftor (LUM), Orkambi, elexacaftor (ELX), tezacaftor (TEZ), CF triple therapy, CF therapy. Articles published between 2014 - 2021 were reviewed.
In CF, the lungs are characterized by chronic inflammation that is driven by the infiltration of immune cells into the airways. The first cells to migrate into the airways are neutrophils. Although neutrophils are recruited to fight bacterial and fungal infections, their activation has the potential to damage surrounding lung tissue by releasing oxidants and protease enzymes [ 6 ]. In both the pediatric and adult populations, the overall cascade results in recurrent lung infections and pulmonary exacerbations which lead to weakened and impaired lungs [ 9 ].
After the discovery of the CF gene in 1989, researchers could figure out how different CFTR mutations cause biochemical and functional abnormalities in the CFTR protein. As a result, research into CFTR dysfunction and CF pathophysiology provided the knowledge needed for the production of pharmacologic compounds that target these various abnormalities. The discovery of the first clinical CFTR modulators was facilitated by the screening of large compound libraries in cell lines expressing different CFTR mutations. Representatives of two classes (potentiators and correctors) of CFTR-directed compounds have become available to treat patients with CF [ 10 ].
Based on their molecular mechanism of action, potentiators and correctors are the two types of FDA-approved CFTR modulators currently available. Potentiators bind to the CFTR protein in the plasma membrane, increasing the CFTR channel's opening frequency and ion conductance. IVA is the only approved CFTR potentiator, and they originally approved it for patients with the G551D mutation. CFTR correctors target the protein-folding defect that results from F508 gene deletion. Until recently, the only clinically approved corrector was LUM [ 11 ].
IVA a CFTR potentiator that enhances the gating frequency of CFTR channels on the cell surface. It was approved to improve the function of mutant CFTR protein. IVA's primary target is a CFTR protein whose glycine at position 551 has been replaced by aspartic acid (G551D). It is recommended to treat patients with multiple CFTR gating mutations (including G551D and other less common non-G551D class III mutations) [ 12 ].
The efficacy and safety of IVA were evaluated in two randomized placebo-controlled trials. Treatment with IVA resulted in a substantial improvement in FEV1 at 24 weeks in both trials, and these improvements persisted at 48 weeks. One trial showed that the mean improvement in percent expected FEV1 from baseline to 24 weeks was 10.6 percentage points higher in the IVA group compared to the placebo group. The other trial showed a 12.5 percentage points higher expected FEV1 in the IVA group compared to the placebo group. Patients who received the drug saw substantial changes in their CF symptoms and were 55% less likely to experience a pulmonary exacerbation than patients who received a placebo [ 13 ]. IVA reduced sweat chloride levels significantly but did not enhance lung function, meaning that a potentiator alone is not enough to save this mutated protein [ 14 ]. Some studies have shown that IVA increased innate immune cell activities, including the killing of Pseudomonas aeruginosa by neutrophils and monocytes [ 8 ].
In 2015, FDA approved the use of Orkambi in patients (aged 12 years or more) who are homozygous for F508del. Orkambi is a fixed-dose tablet containing a combination of LUM and IVA [ 11 ]. Two studies (TRAFFIC) and (TRANSPORT) researched the efficacy of Orkambi, and it showed an improvement in the percent predicted of FEV1 (ppFEV1). LUM-IVA increased ppFEV1 by 2.5%-2.6% (lower bound) to 4.0%-4.1% (upper bound) depending on the dose used [ 8 , 15 ] This combination, however, does not completely restore CFTR protein function and is ineffective in patients with the Phe508del-minimal function (MF) mutation [ 16 ]. Many experts believe that starting CFTR modulator therapy would alter the path of their CF lung disease. Indeed, clinical trials are gradually expanding the use of previously approved drugs to younger age ranges, in the hope that early CFTR modulation can delay or even preclude the development of pulmonary and extrapulmonary complications [ 17 ].
TEZ, the newest CFTR modulator, is a CFTR corrector that was recently approved by the FDA for use in conjunction with IVA. TEZ-IVA combination was found to be efficient in F508del heterozygous patients with a CFTR residual function mutation of the second allele. It enhances F508del-CFTR processing and trafficking. It also increases chloride transport in human bronchial epithelial cells from 2.5% to 8.1% of normal levels. Recent clinical trials have shown that the formulation of TEZ and IVA is marginally superior to its precursor, LUM-IVA, in terms of its improvements to FEV1. The adverse effect profile (including transient bronchoconstriction) and drug-drug interactions are also superior in the TEZ-IVA combination [ 10 , 14 ]
ELX is classified as a next-generation CFTR corrector because it differs from first-generation correctors like TEZ in terms of structure and mechanism. ELX was developed to address the need for an effective CF therapy for patients F508del-CFTR. Patients with genes that do not produce protein or produce proteins that are resistant to IVA or TEZ benefit most from ELX. ELX acts as a CFTR corrector by increasing the amount of mature CFTR proteins on the cell surface. Drugs like ELX help to improve a range of multi-organ CF symptoms, including lung function, nutritional status, and overall quality of life, when used in conjunction with CFTR potentiators, which improve the function of cell-surface CFTR proteins [ 18 ].
In 2019, the FDA approved the use of Trikafta in patients with one copy of F508del-CFTR. Trikafta is the combination of two correctors (TEZ and ELX) and a potentiator (IVA). IVA directly targets mutant CFTR channel-forming protein. TEZ and ELX are thought to also work through direct interaction with mutant F508del-CFTR polypeptide, although evidence is lacking. The mechanism of action of Trikafta is illustrated in Figure Figure1. 1 . They target epithelial cells lining all of the tubular organs affected in CF, including the lungs, gastrointestinal tract, and pancreas. Trikafta costs $311,503 per year, and plans are likely to differ in terms of coverage among insurance companies [ 19 ].

FDA approval of Trikafta came after the results of two randomized, double-blind phase three studies (Trial 1- {"type":"clinical-trial","attrs":{"text":"NCT03525444","term_id":"NCT03525444"}} NCT03525444 and Trial 2- {"type":"clinical-trial","attrs":{"text":"NCT03525548","term_id":"NCT03525548"}} NCT03525548 ) conducted in CF patients aged 12 years and above with at least one F508del mutation [ 20 ]. The first trial was a 24-week placebo-controlled trial that enrolled around 400 patients. The second trial was a four-week active-controlled trial that enrolled around 100 patients with two similar F508del mutations. The trials looked at the pharmacokinetics, safety, and tolerability of ELX/TEZ/IVA over a two-week span. They also assessed the safety, tolerability, efficacy, and pharmacokinetics of the drug over a 24-week treatment period [ 21 ]. The studies showed a significant improvement in lung function, and respiratory-related quality of life, and a decrease in pulmonary exacerbations and sweat chloride after 24 weeks [ 21 ]. Details of the trials mentioned above will be discussed in the next section.
ELX-TEZ-IVA is supplied as a fixed-dose combination tablet of ELX 100 mg, TEZ 50 mg, and IVA 75 mg co-packaged with IVA 150-mg tablets. Adults and children over the age of 12 should administer two fixed-dose combination tablets each morning with a fat-containing meal. The evening dose should be separated by approximately 12 hours from morning administration and consists of one IVA 150-mg tablet taken with a fat-containing meal or snack. This medication is recommended to be taken with foods containing fat. Pancreatic enzymes can also be used for those who are pancreatic insufficient, to maximize efficacy [ 22 ].
1) A study of VX-445 combination therapy in CF subjects homozygous for the F508del (F/F)
This study evaluated the efficacy of VX-445 (ELX) in triple combination (TC) with TEZ and IVA in subjects with CF who are homozygous for the F508del mutation (F/F). The study enrolled 113 participants, of which six participants were included in the run-in period but were not dosed in the TC treatment period. Results were presented for 107 participants dosed in the TC treatment period. The duration of the trial was four months. It was a randomized, double-blind, controlled study. The inclusion criteria included: 12 Years and older CF patients, Homozygous for the F508del mutation (F/F) with Forced expiratory volume in 1 second (FEV1) value ≥40% and ≤90% of predicted mean for age, sex, and height. The two arms of the study were 52 patients treated with TEZ/IVA and 55 patients treated with VX-445/ TEZ/IVA. The primary outcome measured was the absolute change in ppFEV1.
The results showed that the change in ppFEV1 from baseline to four weeks was 10.4 percentage points for the triple therapy group and 0.4 for the TEZ/IVA arm (p<0.0001). Among the secondary outcomes measured, the absolute change in Sweat Chloride (SwCl) at week 4 was -43.4 in the treatment group vs 1.7 in the placebo group (p<0.0001). The absolute change in Cystic Fibrosis Questionnaire-Revised (CFQ-R) Respiratory Domain Score From baseline at week 4 was 16 units in those receiving triple therapy and -1.4 units in the placebo group (p<0.0001).
In terms of adverse events, no death was noted in both groups. Among the serious adverse events, one out of 55 patients from the triple therapy arm was diagnosed with infective pulmonary exacerbation of CF while one out of 52 patients experienced the same adverse event in the placebo group. Both groups reported diarrhea, abdominal pain, nausea, headache, cough, and fatigue [ 23 ].
2) A phase 3 study of VX-445 combination therapy in subjects with CF heterozygous for the F508del mutation and a minimal function mutation (F/MF)
This study evaluated the efficacy of VX-445 in triple combination with TEZ and IVA in subjects with CF who are heterozygous for F508del and a minimal function mutation (F/MF subjects). A total of 405 participants were enrolled in the study, of which two participants were enrolled but were not dosed in the triple combination treatment period. Results are presented for 403 participants dosed in the TC treatment period. The duration of the trial was 10 months. It was a randomized, double-blind, controlled trial. Inclusion criteria included: 12 Years and older CF patients, Heterozygous for the F508del mutation (F/MF) with FEV1 value ≥40% and ≤90% of predicted mean for age, sex, and height. The two arms were the placebo group and the treatment group receiving VX-445/TEZ/IVA. The primary outcome measured was the absolute change in ppFEV1.
The results showed a change in ppFEV1 from baseline at Week 4 was 13.6 percentage points for the treatment group and -0.2 for the placebo group (p<0.0001). Among the secondary outcomes measured, the absolute change in ppFEV1 from baselines at week 24 was 13.9 percentage points for the treatment group and -0.4 for the placebo group (p<0.0001).
The number of pulmonary exacerbations (PEx) from baseline at week 24 was 41 for the treatment group and 113 for the placebo group (p<0.0001). The absolute change in sweat chloride from baseline at Week 24 was -42.2 in the treatment group and -0.4 in the placebo group (p<0.0001). The absolute change in the CFQ-R respiratory domain score from Baseline at Week 24 was 17.5 points for the treatment group and -2.7 for the placebo group (p<0.0001). The absolute change in body mass index (BMI) from Baseline at Week 24 was 1.13 in the treatment group and 0.009 in the placebo group.
In terms of AE, no significant differences were found between groups in terms of adverse effects. No death was reported. 13.8 % of patients in the VX-445/TEZ/IVA group experienced serious side effects (intestinal obstruction, respiratory tract infections, rash, etc.) vs 20.9% in the placebo group. Common noted not-serious adverse effects include diarrhea, abdominal pain, vomiting, and fatigue [ 24 ].
3) Evaluation of VX 445/TEZ/IVA in CF subjects 6 through 11 years in age
This study evaluated the pharmacokinetics, safety, tolerability, efficacy, and pharmacodynamic effect of VX-445, TEZ, and IVA when dosed in TC in CF subjects 6 through 11 years of age. The study included 66 participants. Inclusion criteria were: CF patients aged 6 to 11 years, homozygous or heterozygous for F508del mutation, and FEV1 value ≥40% of predicted mean for age, sex, and height. The study was completed on August 7, 2020. However, the results are yet to be published [ 25 ].
The discovery of the CFTR gene in 1989 paved the way for researchers to learn more about the structure, processing, and role of CFTR in health, revealing how multiple mutations in this epithelial anion channel cause a multiorgan disease. The approval of ivacaftor as the first CFTR modulator drug was a significant step forward and an important proof-of-concept for causal pharmacotherapy for this life-shortening genetic disorder on a larger scale. This CFTR potentiator restored around 50% of the CFTR function and showed improvement in lung function. And after the approval of the first corrector, lumacaftor combination therapy was used, and it was superior to monotherapy. Recent early phase clinical trials show that using triple-combination therapies with a second corrector compound is necessary to repair multiple defects in F508del processing. CF newborn screening has provided an opportunity to take advantage of the modulators in infants and young children, which have the potential to postpone or even avoid irreversible structural lung damage. However, CFTR modulator therapies may not help approximately 10% of the CF population.
The CFTR modulators IVA, LUM/IVA, TEZ/IVA, and ELX/TEZ/IVA have been shown to be effective not just in people with mild to severe CF, but even in people with advanced pulmonary disease, such as lung transplant applicants. Randomized controlled trials and open-label studies, which enrolled participants with more severe pulmonary disease than those inadvertently included in the randomized controlled trials, strongly showed this beneficial effect. It is widely accepted that the most significant effect of CFTR modulator therapy can be seen when it is initiated as early as possible in the disease's course when permanent lung damage is at the least serious. With the latest approval of the triple combination treatment for most people with CF by the FDA, we should expect more people with serious CF pulmonary disease to benefit from CFTR modulation.
Conclusions
Trikafta is expected to cut the number of prescriptions prescribed every day in half and reduce the time spent on therapy per day. This will then cut down on regular care time, total cost, and treatment stress for people dealing with CF. Perhaps future experiments will prove this theory. Trikafta was revolutionary in improving the day-to-day activities of CF patients. A member of a support group on cysticfibrosis.com summarized her life-changing experience. She tried the Trikafta treatment for 12 months. She divided the treatment into two parts. Part 1 was the first six months. She referred to the treatment as a "silver bullet.” She noticed so many changes in her body and in her daily life because of Trikafta, ranging from deeper breaths to having more time and energy to lie on the floor and play blocks with her daughter. However, her major change was that she could talk on the phone while walking without becoming short of breath. In addition, she reported that during part 1, she had only one mild pulmonary exacerbation and it did not require hospital admission. Her PFT was increased by 10% after three months of treatment. Her cough vanished. Shortly after starting Trikafta, her 7% sodium chloride nebulizer treatment felt too strong. Her doctor diluted the treatment from 7% to 3.5% solution because the lungs were becoming too clear to have such a strong treatment. In part 2, she reported skin and nail changes, decreased joint and body aches, increased energy, and an increase in her appetite without nausea. This drug was truly magical in improving her lifestyle.
Additional studies for greater intervention groups and longer intervention times may be conducted to better show the treatment's potential in curing the underlying protein deficiency. For example, the clinical trial with CF patients less than 12 years old has been completed recently and results will be published soon. This treatment can avoid multi-organ manifestations of the disease and could enable young children with CF to live a normal life span. Implementing Trikafta is expected to lead to significant changes in the lives of people with CF. This level of CFTR modulation in such a large proportion of CF patients could have a significant impact on CF treatment.
The content published in Cureus is the result of clinical experience and/or research by independent individuals or organizations. Cureus is not responsible for the scientific accuracy or reliability of data or conclusions published herein. All content published within Cureus is intended only for educational, research and reference purposes. Additionally, articles published within Cureus should not be deemed a suitable substitute for the advice of a qualified health care professional. Do not disregard or avoid professional medical advice due to content published within Cureus.
The authors have declared that no competing interests exist.
IMAGES
COMMENTS
The cystic fibrosis transmembrane conductance regulator (CFTR) is an anion channel that regulates salt and fluid homeostasis across epithelial membranes 1. Alterations in CFTR cause cystic ...
2.1. Characteristics of the human cystic fibrosis gene and encoded CFTR protein. Cystic fibrosis is caused by pathogenic mutations in a single large gene located on human chromosome 7 that encodes the cystic fibrosis transmembrane conductance regulator (CFTR) protein. 1, 2, 3 CFTR belongs to the ABC (ATP‐binding cassette) family of proteins, a large group of related proteins that share ...
Cystic fibrosis is a rare genetic disease caused by mutations in CFTR, the gene encoding cystic fibrosis transmembrane conductance regulator (CFTR). The discovery of CFTR in 1989 has enabled the ...
1. Introduction. Cystic Fibrosis (CF) is a lethal recessive disease caused by loss-of-function mutations in the CFTR (cystic fibrosis transmembrane conductance regulator). CF frequently occurs among Caucasians (about 1 in 3500), and the number of patients is more than 90,000 world-wide (US: approximately 30,000, Europe: approximately 48,000) [].CF patients present with progressive disease ...
Following discovery of the cystic fibrosis transmembrane conductance regulator (CFTR) gene in 1989 and subsequent elucidation of the varied CFTR protein abnormalities that result, a new era of cystic fibrosis management has emerged—one in which scientific principles translated from the bench to the bedside have enabled us to potentially treat the basic defect in the majority of children and ...
Biallelic mutations of the cystic fibrosis (CF) transmembrane regulator (CFTR) gene resulting in reduced or absent protein function cause CF. 1 The observed clinical syndrome of CF centers on airway disease characterized by inflammation, chronic infections, inspissation of tenacious secretions, bronchiectasis, and eventually premature death.This syndrome has been extensively reviewed. 2, 3 ...
Hisert, K. B. et al. Restoring cystic fibrosis transmembrane conductance regulator function reduces airway bacteria and inflammation in people with cystic fibrosis and chronic lung infections. Am. J.
The landscape of cystic fibrosis care has transformed since the discovery of the cystic fibrosis transmembrane regulator (CFTR) gene defect more than 30 years ago.Due to the collaborative efforts of the global cystic fibrosis community in both research and clinical care, the focus is now on disease modification with highly effective medications rather than on solely managing the downstream ...
Substances. Cystic Fibrosis Transmembrane Conductance Regulator. Chronic obstructive pulmonary disease (COPD) manifests with a variety of clinical presentations, reflecting its complex pathology. Currently, care focuses on symptom amelioration and prevention of complications and thus is generally tailored to disease severity rather than ...
Cystic fibrosis transmembrane conductance regulator (CFTR) is a cAMP- and protein kinase A (PKA)-regulated channel, expressed on the luminal surface of secretory and absorptive epithelial cells. ... miR-223, miR-494, and miR-509, have been repeatedly reported by several research groups as direct inhibitors of CFTR mRNA [28], [29 ...
The cystic fibrosis transmembrane conductance regulator (CFTR) gene has been traditionally linked to cystic fibrosis (CF) inheritance in an autosomal recessive manner. Advances in molecular biology and genetics have expanded our understanding of the CFTR gene and its encoding products expressed in different tissues. The study's aim consists of reviewing the different pathological CF ...
Introduction. Cystic fibrosis is one of the most common inherited diseases, with roughly 1 in 22 carriers in the Caucasian population and with an incidence of about 1 in 2500 live births. The primary defect is loss of function of a plasma membrane-located chloride channel termed the cystic fibrosis transmembrane conductance regulator (CFTR) 2.
Introduction. Cystic fibrosis (CF) is a classical autosomal recessive genetic disease, caused by loss-of-function mutations in a single gene, that which encodes the cystic fibrosis transmembrane conductance regulator (CFTR) [].CFTR is a member of a large family of membrane transport proteins, the ATP-binding cassette (ABC) family, which is comprised of 48 members in humans subdivided into ...
Cystic fibrosis (CF) is a fatal autosomal recessive disorder caused by the loss of function mutations within a single gene for the Cystic Fibrosis Transmembrane Conductance Regulator (CFTR). CFTR is a chloride channel that regulates ion and fluid transport across various epithelia. The discovery of CFTR as the CF gene and its cloning in 1989, coupled with extensive research that went into the ...
The cystic fibrosis transmembrane conductance regulator (CFTR) is a member of the ATP-binding cassette (ABC) transporter superfamily. CFTR controls the flow of anions through the apical membrane of epithelia. Dysfunctional CFTR causes the common lethal genetic disease cystic fibrosis. Transitions between open and closed states of CFTR are ...
The emerging use of cystic fibrosis (CF) transmembrane conductance regulator (CFTR) modulators has provided CF patients with unprecedented access to potentially life-extending therapies 1.In 2015 ...
This work was supported by the Canadian Institutes for Health Research and Alberta Innovates Technology Futures. The authors declare that they have no conflicts of interest with the contents of this article.This article contains supplemental sequences and structures.The abbreviations used are:CFTRcystic fibrosis transmembrane conductance regulatorTMDtransmembrane domainABCATP-binding cassetter ...
Cystic fibrosis (CF) is the most frequent lethal genetic disease in the Caucasian population. CF is caused by a defective gene coding for the cystic fibrosis transmembrane conductance regulator (CFTR), a cAMP- and ATP-dependent Cl - channel and central regulatory protein in epithelia. CFTR influences the fluid composition of the mucus in the ...
Abstract. The cystic fibrosis transmembrane conductance regulator (CFTR) is responsible for the disease cystic fibrosis (CF). It is a membrane protein belonging to the ABC transporter family functioning as a chloride/anion channel in epithelial cells around the body. There are over 1500 mutations that have been characterised as CF-causing; the ...
Cystic fibrosis (CF) is caused by the functional expression defect of the cystic fibrosis transmembrane conductance regulator (CFTR) protein. Despite the recent success in CFTR modulator development, the available correctors only partially restore the F508del-CFTR channel function, and several rare CF mutations show resistance to available drugs. We previously identified compound 4172 that ...
Cystic fibrosis is an autosomal recessive, monogenetic disorder caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene. The gene defect was first described 25 ...
Gene transfer could provide a novel therapeutic approach for cystic fibrosis (CF), and adeno-associated virus (AAV) is a promising vector. However, the packaging capacity of AAV limits inclusion of the full-length cystic fibrosis transmembrane conductance regulator (CFTR) cDNA together with other regulatory and structural elements.
Relationships between cystic fibrosis transmembrane conductance regulator, extracellular nucleotides and cystic fibrosis. by Brice Marcet, Jean-Marie Boeynaems. Pharmacology & therapeutics. Read more related scholarly scientific articles and abstracts.
Cystic fibrosis (CF) is a recessive genetic disease caused by a mutation in the epithelial chloride channel—cystic fibrosis transmembrane conductance regulator (CFTR). CF is a predominant genetic disorder with a disease severity ranging from mild to life-threatening. The number of CF patients in a population varies depending on ethnicity.
The cystic fibrosis transmembrane conductance regulator (CFTR) protein helps to maintain the balance of salt and water on many surfaces in the body, such as the surface of the lung. The CFTR protein is a particular type of protein called an ion channel. In the lung, the CFTR ion channel moves chloride ions from inside the cell to outside the cell.
Introduction and background. Cystic fibrosis (CF) is a chronic, progressive, autosomal recessive disease that affects approximately 35,000 people in the United States [1,2].The primary defect is a mutant CFTR gene causing a decrease or absence of cystic fibrosis transmembrane conductance regulator (CFTR) activity.